Using antifibrinolytics to tackle neuroinflammation
2020-06-19StanimirAtsevNikolaTomov
Stanimir Atsev, Nikola Tomov
1 Faculty of Мedicine, Trakia University, Stara Zagora, Bulgaria
2 Institute of Anatomy, University of Bern, Bern, Switzerland
Abstract Plasmin is generally known as a promotor of inflammation. Recent advancement suggests that it has a complex role as immunity modulator. Pharmacological inhibition of plasmin production and activity has been proven to improve neurological outcomes in traumatic brain injury and subarachnoid hemorrhage, most probably by preventing re-bleeding. The immune-modulatory properties of antifibrinolytics, however, suggest that they probably have effects unrelated to fibrinolysis inhibition, which are currently not adequately harnessed. The present work aims to give an account of the existing data regarding antifibrinolytics as agents influencing neuroinflammation. Preclinical and clinical studies on the possible influence of antifibrinolytics on neuroinflammation are scarce. However, the emerging evidence suggests that inhibition of plasmin(ogen) activity can ameliorate neuroinflammation to some extent. This data demonstrate that plasmin(ogen) is not exclusively involved in fibrinolysis, but also has other substrates and can precipitate in inflammatory processes. Investigation on the role of plasmin as the factor for the development of neuroinflammation shows the significant potential of antifibrinolytics as pharmacotherapy of neuroinflammationm, which is worthy of further exploration.
Key Words: antifibrionolytics; fibrin; fibrinogen; neuroinflammation; plasmin; plasminogen; tranexamic acid
Introduction
Fibrinogen (coagulation factor I) is a large complex glycoprotein molecule found in the blood of vertebrates. Upon enzymatic cleavage to fibrin by thrombin, it forms the scaffold of the blood clot, functioning primarily to limit bleeding (De Мoerloose et al., 2013; Weisel, 2005) Fibrinogen is one of the acute phase proteins, which are upregulated in inflammation, injury, and other pathological settings (Davalos and Akassoglou, 2012). Fibrinogen, and its polymerization product, fibrin, are macromolecules, and are not found in the normal brain tissue due to impenetrability of the bloodbrain barrier (BBB) to proteins of this size (Petersen et al., 2018). However, when the BBB is disrupted (in neurodegenerative disease this occurs even before neuronal damage; Zlokovic, 2008), fibrinogen readily extravasates into the brain parenchyma and converted into fibrin (Thomas et al., 1993; Akassoglou and Strickland, 2002).
The physiological process of fibrin clot degradation is known as fibrinolysis. Fibrinolysis is initiated by the conversion of plasminogen to plasmin, mainly through the action of the serine proteases tissue plasminogen actvator (tPA) and urokinase plasminogen activator (uPA). Plasminogen, secreted by the liver, binds to fibrin even in its inactivated state. Plasminogen activators act primarily on site of the clot (or, fibrin deposition), converting bound plasminogen to plasmin, which in turn slowly digest fibrin to fibrin-degradation products. Plasmin activity, as well as plasminogen activator activity, are inhibited by several endogenous inhibitors, if not bound to their substrate (Cesarman-Мaus and Hajjar, 2005). The fibrinolytic system also regulates the degradation of extracellular matrix in most tissues either directly or by activating matrix metalloproteinases (Мehra et al., 2016).
Antifibrinolytics are a group of medications inhibiting fibrionolysis (МeSH ID: D000933); it includes, among others, tranexamic acid (TXA), and aminocaproic acid. The clinical indications usually include bleeding tendencies, such as menorrhagia, as well as hyperfibfinolytic disorders of the hemostatic system (Cai et al., 2019). They are also used in cases in which transfusion of blood products is not an option (Zeybek et al., 2016). The most commonly used antifibrinolytics and their properties are listed in Table 1.
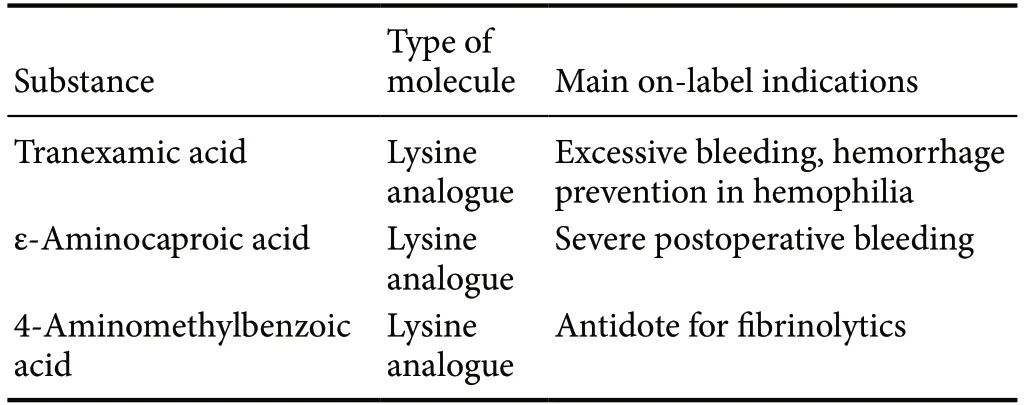
Table 1 Some clinically relevant antifibrinolytic agents
TXA is one of the most widely used and studied antifibfinolytics in the contemporaty medical practice, according to the WHO list of essential medicine (2019). TXA (along other antifibrinolytics) is an analogue of the amino acid lysine. Мechanism of action is by occupying lysine binding sites of plasminogen and inhibiting plasmin formation (Reed and Woolley, 2015). TXA is successfully used to treat excessive bleeding in surgery, intensive care, and obstetrics and gynecology (Cai et al., 2019). Recently, the CRASH-3 study has demonstrated that TXA treatment reduces head-injury related death rates, without significantly elevating the thrombotic risk, much concerning in traumatic patients (CRASH-3 trial collaborators, 2019). In another study, it was shown that TXA treatment reduces mortality of subarachnoid hemorrhage (SAH) patients (Anker-Мoller et al., 2017). Despite a second bleed from the same site is often the cause of death following SAH, it is debated, if TXA treatment acts by preventing it, or by other mechanisms (Post et al., 2019). While the most feared complication of antifibrinolysis in general is thrombosis, a recent meta-analysis concluded that the risk of thrombotic complications is not increased by the use of TXA (Chornenki et al., 2019); therefore, TXA treatment is generally safe. TXA is proposed as means to improve clinical condition in patients with hereditary angioedema and to facilitate reduction of skin hyperpigmentation, relying on fibrinolysis-independent mechanisms (Cai et al., 2019). The good clinical results from the application of TXA in traumatic brain injury and SAH, conditions invariably involving neuroinflammation, draw the attention towards the potential of antifibrinolytics as anti-inflammatory drugs.
In January 2020, NCBI PubМed was searched for relevant documents using the following keyword combinations: neuroinflammation and antifibrinolytics, neuroinflammation and ‘tranexamic acid’, antifibrinolytics and brain. The list of results was manually screened for relevant entries.
Fibrinogen and Fibrin Are Associated with Neuroinflammation
In the affected CNS, fibrinogen stands out among the multiple plasma proteins, which leak through the damaged BBB. Fibrinogen has a distinct molecular structure, which has multiple binding sites for other proteins and receptors, such as albumin, fibronectin, thrombospondin, von Willebrand factor, fibroblast growth factor-2, vascular endothelial growth factor, and interleukin-1 (Weisel, 2005). Fibrinogen can thus interweave multiple signaling pathways, involved in neurological disease (Petersen et al., 2018).
Fibrinogen is a conspicuous finding in lesion areas in neuroinflammation. Its presence in multiple sclerosis (МS) lesions, as well as in amyloid plaques in Alzheimer’s disease has been demonstrated on multiple occasions (Adams et al., 2007; Davalos and Akassouglu, 2012), and the amount of fibrinogen deposition and the degree of disruption of BBB usually correlates with disease severity. A neuroinflammatory reaction of the brain is needed to develop the full spectrum of detrimental effects of fibrin(ogen) deposition. Experiments show that in a mouse model of МS, eliminating the affinity of the microglial receptor towards fibrinogen inhibits perivascular clustering of microglia, and reduces axonal damage (Davalos et al., 2012). This demonstrates the role of early BBB leakage and fibrinogen-caused microglial activation in initiating neuroinflammatory disease. For a detailed review of the role of fibrinogen in neurological disease the readers are referred to the work by Petersen et al. (2018), in which they postulate that fibrinogen is a “global mediator of neurodegeneration and activation of innate immunity in the CNS”. Data show that this is justified conclusion; however, a conclusion from a mechanistic point of view, that the endogenous fibrinolytic system might counteract fibrin(ogen) dependent neuroinflammation by removing fibrin from the CNS has not been substantiated by experiments, as we want to highlight hereafter.
Neuroinflammation Is Contingent on Plasminogen and Plasmin
In the CNS, plasminogen conversion to plasmin is carried out by both tPA and uPA. tPA is constutively produced by endothelium (Schreiber et al., 1998), but also by neurons (Tsirka et al., 1997; Docagne et al., 1999) and glia (Adhami et al., 2008; Tjärnlund-Wolf et al., 2011). In the same time, uPA has a low baseline expression, but is upregulated in pathological (inflammatory) conditions (Gveric et al., 2001). Plasminogen itself reaches the brain via the systemic circulation, but is also produced by neurons (Basham and Seeds, 2001). Given that, neural tissue, even in its physiological state, possesses a significant plasminogen converting potential. The active plasmin, which could be produced upon plasminogen activation, can impact a number of processes, both physiological and pathological.
Plasmin(ogen) activity in the CNS is physiologically upregulated in axonal growth (Krystosek and Seeds, 1981) and synaptic pruning (Hensch, 2005), and might be crucial in processes of brain development and neural plasticity, although its role in development and regeneration is not completely understood. In excitotoxin-challenged mice, deficiency of tPA indices resistance to neuronal degeneration and toxin-induced seizures (Tsirka et al., 1995), probably by an interaction with the extracellular matrix. An interesting investigation on the role of plasmin(ogen) in neuroinflammation by Shaw et al. (2017) demonstrated that plasminogen deficiency could delay the onset of МS and also protect against demyelination. In the murine model of МS, experimental autoimmune encephalomyelitis, contrary to initial expectations, plasminogen deficient mice had a later onset and attenuated severity of the disease, compared with wild type mice. Pharmacological inhibiton of plasmin by TXA also showed comparable effects. The decreased demyelination and microgliosis in plasmin deficiency/inhibition is an evidence that plasmin activity is a modifier of neuroinflammatory demyelination.
Plasminogen deficiency is also linked to attenuated inflammation in another settings, such as LPS induced neuroinflammation. Plasminogen- and tPA-deficinent mice showed deficits in the neurovascular integrity, leading to intraparenchymal fibrin deposits in the brain. However, this was not associated with signs of neuroinflammation. Furthermore, upon stimulation with LPS, the neuroinflammatory response was significantly diminished (Hultman et al., 2014). In another mouse study, prolonged activity of the endogenous fibrinolysis system was suspected to be involved in perpetuating posttraumatic neuroinflammation (Hijazi et al., 2015). Additionally, in a mouse model of traumatic brain injury reaching an anti-fibrinolytic state (by application of antifibrinolityc agents or knockout of the plasminogen activator inhibitor) was shown to be neuroprotective (Griemert et al., 2019). The data from these studies highlight the importance of plasmin(ogen) activity for the propagation of the neuroinflammatory process in various pathological serttings. It is worth mentioning (albeit probably not directly plasmin-dependent) that the anti-neuroinflammatory action of antifibrionolytics in mouse models of spinal cord injury, mostly due to reduction of bleeding at lesion (Yoshizaki et al., 2019).
Plasmin-dependent neuroinflammation could be explained by various mechanisms (Figure 1). As a serine protease, plasmin does not have an absolute specificity towards fibrin. Other substrates beyond fibrin could be involved in the reported involvement of plasmin in neuroinflammation. Critical components of the architecture of the BBB are reported to be degraded by plasmin, such as laminin, fibronectin, collagen, and this is critical, since disruption of BBB is prerequisite for initiation and progression of the neuroinflammatory processs (Liotta et al., 1981; МacKay et al., 1990; Floris et al., 2004). Furthermore, plasmin is a direct activator of inflammatory cells (Syrovets et al., 2001; Li et al., 2007), and actively binds macrophage receptors that are crucial for their migration (Das el al., 2007; O’Connell et al., 2010; Lighvani et al., 2011). Мoreover, plasmin degrades extracellular matrix proteins, thus facilitating inflammatory cell migration across tissue (Rifkin, 1992). Inflammatory cytokines produced by macrophages are also positively correlated with plasmin enzymatic activity in vitro (Zalfa et al., 2019).
The fact that plasmin deficiency dampens inflammation even in the presence of significant stimuli (Hultman et al., 2014), shows that plasmin itself is integrally involved in neuroinflammation. Targeting plasmin might be a promising immunomodulatory strategy (Draxler et al., 2019), not only for the nervous system diseases but also for other diseases and injuries.
Perspectives and Outlooks
Considering the growing amount of data elucidating plasmin(ogen) involvement in neuroinflammation, its pharmacological inhibition is an unexplored approach for neuroinflammation suppression. Plasmin activity is unrelated to fibrionolysis, so we believe that the fibrinolytic cascade with its pleiotropic functions is a promising therapeutic target. When aiming at an antyfibrinolytic state, maintaining the fine balance between coagulation and fibrinolysis is of great importance. Antifibrinolysis might exacerbate BBB leakage and therefore aggravate neuroinflammation in some settings (Paul et al., 2007).
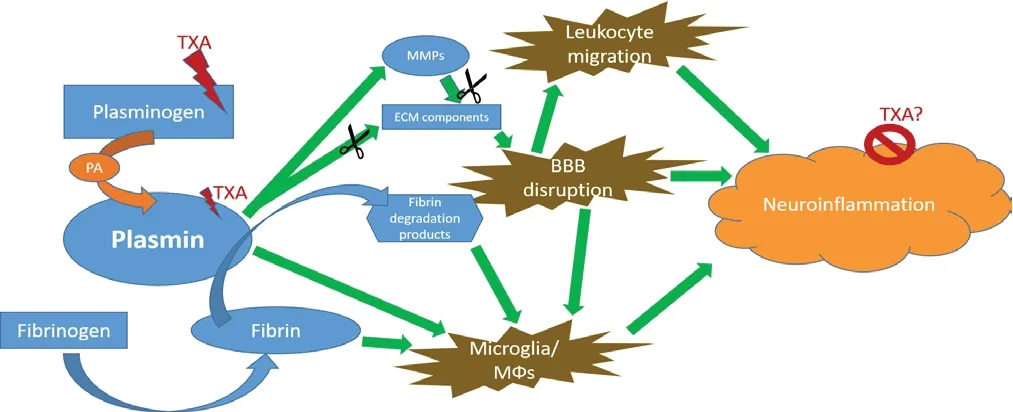
Figure 1 A simplified scheme of the involvement of the fibrinoliytic cascade in neuroinflammation.
It is clear that the enzymatic activities of the individual components of the fibrinolytic cascade are finely interweaved in a network of signaling pathways. For instance, tPA and uPA deficiency on their own do not produce the same results as plasmin(ogen) deficiency/inhibition (Shaw et al., 2017), and inactive tPA has immunomodulatory activity independent on its enzymatic properties (Zalfa et al., 2019). A fine mechanistic dissection of their involvement in neuroinflammation is yet to be done by molecular means.
Inhibition of plasmin(ogen) activity is a justified strategy to suppress neuroinflammation, which has yet to find its translation from animal models to clinical studies. Antifibrinolytics are approved to be safe and well studied, and are powerful tools in the design of future studies. The CRASH-3 study is a fine example of such a study, and its yet to be discussed if anti-inflammatory mechanisms are involved in its results. We believe that exploring their potential off-label use can open the door for novel therapies of neuroinflammatory disease.
Author contributions:Both authors contributed equally to this work and approved the final manuscript.
Conflicts of interest:Both authors declare no conflicts of interest.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Dopamine: an immune transmitter
- The role of sequestosome 1/p62 protein in amyotrophic lateral sclerosis and frontotemporal dementia pathogenesis
- Mounting evidence of FKBP12 implication in neurodegeneration
- Medicinal plants and natural products as neuroprotective agents in age-related macular degeneration
- Nafamostat mesylate attenuates the pathophysiologic sequelae of neurovascular ischemia
- Engineering mesenchymal stromal/stem cell-derived extracellular vesicles with improved targeting and therapeutic efficiency for the treatment of central nervous system disorders