盐酸帕罗西汀肠溶缓释片的研制及体外释放度和体内药物动力学评价
2020-06-18蔡燕霞
蔡燕霞, 程 刚
盐酸帕罗西汀肠溶缓释片的研制及体外释放度和体内药物动力学评价
蔡燕霞, 程 刚*
(沈阳药科大学 药学院, 辽宁 沈阳 110016)
研制盐酸帕罗西汀肠溶缓释片,研究其体外释放及人体药物动力学参数。以羟丙甲纤维、山嵛酸甘油酯为缓释材料,用双层压片及肠溶包衣工艺制备缓释片。以高效液相色谱法进行体外释放测试,采用2相似因子法评价自制制剂和参比制剂的体外释放曲线,并研究了参比制剂及两批不同处方的自制制剂在14名健康志愿者中的单剂量药物动力学。两个自制制剂和参比制剂体外释放试验2相似因子大于50,相对生物利用度分别为108.70% 和146.82%。研制的盐酸帕罗西汀肠溶缓释片具有缓释特性,其中一个自制制剂在扩大样本量后可达到和参比制剂生物等效,另一个和参比制剂无法生物等效。体外释放和体内吸收具有一定的相关性,但2结果不能反映盐酸帕罗西汀肠溶缓释片的体内生物等效性。
盐酸帕罗西汀肠溶缓释片; 体外释放; 药物动力学;2相似因子法
盐酸帕罗西汀属于5-羟色胺再摄取抑制剂(SSRIs),用于治疗各种类型的抑郁症,包括伴有焦虑的抑郁症及反应性抑郁症[1-4]。盐酸帕罗西汀通过竞争性抑制神经元突触前膜对5-羟色胺(5-HT)的再摄取,使突触间隙中5-羟色胺浓度升高,有利于5-羟色胺能神经的冲动传递,增强中枢5-羟色胺神经功能。盐酸帕罗西汀无镇静作用,正常剂量对心率、血压无影响,对胆碱能、组胺或肾上腺能受体的亲合力低,抗胆碱性、心血管的不良反应小于三环类抗抑郁剂,选择性较氟西汀、舍曲林及氯丙咪嗪强[1-4]。1999年2月16日,盐酸帕罗西汀肠溶缓释片经FDA批准上市,商品名为Paxil CR,规格为12.5 mg、25 mg和37.5 mg,缓释片较速释片血药浓度波动小,不良反应的发生率也明显降低。早期文献报道,盐酸帕罗西汀肠溶缓释片均为单层片制备工艺[5-8],多采用羟丙甲纤维制备成亲水凝胶骨架片后对释放曲线的相似性进行研究,并无体内药物动力学的相关报道。原研制剂Paxil CR为双层片,含药层为含有羟丙甲纤维的亲水凝胶骨架层,非含药层为含有山嵛酸甘油酯的溶蚀层[9]。本文作者采用双层片制备工艺制备盐酸帕罗西汀肠溶缓释片,以释放度曲线为指标,对双层片处方工艺的影响因素进行考察,并制备不同释放曲线的自制制剂,以原研制剂为参比制剂,采用随机、开放、三制剂、三周期交叉试验设计进行单剂量药物动力学实验,旨在对盐酸帕罗西汀肠溶缓释片的体外释放及人体内药物动力学进行研究,评价其体内外相关性,并预测所开发的自制制剂的生物等效性。
1 仪器与试药
ZPW23旋转式双层压片机(上海天祥健台制药机械有限公司),BGB-5F实验型高效包衣机(浙江小伦制药机械有限公司),HLY-10混合制粒机(上海天祥健台制药机械有限公司),DPL-II多功能制粒包衣机(重庆精工制药机械有限责任公司),HD-20B多向运动混合机(浙江小伦制药机械有限公司),Agilent 708-DS自动溶出仪(美国Agilent公司),岛津 UV-1700紫外分光光度计(日本岛津公司),盐酸帕罗西汀(惠州信立泰药业有限公司),乳糖(DMV-Fonterra Excipients GmbH & Co.KG),羟丙甲纤维素K4M(陶氏化学公司),羟丙甲纤维素E5(陶氏化学公司),山嵛酸甘油酯(GATTEFOSSE SAS),聚维酮K30(BASF Corporation),甲基丙烯酸-丙烯酸乙酯共聚物水分散体(Evonik Nutrition & Care GmbH),硬脂酸镁(湖州展望药业有限公司),二氧化硅(安徽山河药用辅料股份有限公司),滑石粉(广西龙胜华美滑石开发有限公司),柠檬酸三乙酯(蚌埠丰原涂山制药有限公司),其他试剂为分析纯。
2 方法与结果
2.1 缓释片制备方法
2.2.1 片芯的制备
将含药层处方量盐酸帕罗西汀、乳糖、羟丙甲纤维素(K4M)加入至湿法制粒机混合均匀,加入质量分数5%的聚维酮K30溶液制粒,流化床55 ℃干燥,24目筛整粒,加入处方量硬脂酸镁、二氧化硅混合均匀,得含药层颗粒。将阻挡层山嵛酸甘油酯、乳糖、羟丙甲纤维素(E5)、红氧化铁加入至湿法制粒机内混合均匀,加入质量分数5%的聚维酮K30溶液制粒,流化床55 ℃干燥,24目筛整粒,加入处方量硬脂酸镁、二氧化硅混合均匀,得阻挡层颗粒。将含药层颗粒及阻挡层颗粒用双层压片机压制成双层片,控制片剂硬度为4~7 kgf。
2.2.2肠溶衣的包制
将处方量甲基丙烯酸-丙烯酸乙酯共聚物水分散体、枸橼酸三乙酯及滑石粉加入至纯化水中,搅拌均匀;将片芯置高效包衣锅中,设置包衣锅转速为4~6 r·min-1,片床温度为30~50 ℃,雾化压力为0.15 MPa,包衣液流量,干燥时间为2 h,增重适量。
2.2 方法学考察
2.2.1 对照溶液的制备
取盐酸帕罗西汀对照品适量,精密称定,加甲醇溶解并定量稀释制成每1 mL中约含0.5 mg的溶液,作为对照品溶液。
2.2.2 释放度介质的配制
0.1 mol·L-1盐酸:取盐酸9 mL,加已脱气水稀释至1 000 mL,混匀,既得。
pH 6.8缓冲液:取磷酸二氢钾6.8 g、氢氧化钠0.9 g,加已脱气水稀释至1 000 mL,用氢氧化钠调节pH值至6.8,混匀,即得。
pH 7.5缓冲液:取三羟甲基氨基甲烷6.06 g,加已脱气水稀释至1 000 mL,用盐酸调节pH值至7.5,混匀,即得。
2.2.3 色谱条件
检测波长:295 nm,色谱柱:三甲基硅烷键合硅胶柱,流动相:乙腈-醋酸铵缓冲液-三乙胺(体积比40︰60︰1),用冰醋酸调节pH值至5.5,流速:1.0 mL·min-1,进样量:10 μL。
2.2.4 标准曲线
配制含量相当于供试品溶液浓度25%、50%、100%、120%、150%、200%的一系列溶液。精密量取上述溶液各10 μL,注入液相色谱仪,记录色谱图。按最小二乘法进行线性拟合。线性方程为= 6.862 858 927×106+ 3.732 556 15×104,= 0.9999,结果表明,帕罗西汀在0.111 5~0.891 6 g·L-1,相当于100%供试品浓度的22.29%~178.3%范围内线性良好。
2.2.5 精密度试验
取6份不同介质的自制制剂供试品溶液,测定供试品溶液含量,考察精密度,结果见表1。结果表明,本试验方法精密度符合要求。

Table 1 Precision results of paroxetine hydrochloride in different dissolution media
2.2.6 回收率试验
按0.1 mol·L-1盐酸释放度测定方法,精密量取50%回收率供试液、100%回收率供试液、150%回收率供试液共9份溶液各100 μL注入液相色谱仪,按pH 6.8、pH 7.5缓冲液释放度测定方法,精密量取10%回收率供试液、50%回收率供试液、100%回收率供试液、120%回收率供试液共12份溶液各10 μL注入液相色谱仪,考察帕罗西汀的回收率。结果见表2。结果表明,本试验方法回收率符合要求。

Table 2 Recovery results of paroxetine hydrochloride in different dissolution media
2.2.7 稳定性试验
取0.1 mol·L-1盐酸介质的供试品溶液,于0、4、8、12、25、26、49、73、97、121 h时间点进样测定含量,取pH 6.8、pH 7.5介质的供试品溶液于0、5、8、12、27、49、74、97、123 h时间点进样测定含量,结果表明,盐酸帕罗西汀在三种溶出介质中放置74 h内稳定。
2.3 释放度测定
参考《美国药典》中盐酸帕罗西汀肠溶缓释片释放度测定方法[10],取本品,照溶出度与释放度测定法(《中华人民共和国药典》2015年版四部通则0931第一法)[11],以0.1 mol·L-1盐酸溶液750 mL为溶出介质,转速为100 r·min-1,依法操作,2 h时,取溶液适量,滤过,取续滤液作为供试品溶液;另精密称取盐酸帕罗西汀对照品适量,加入甲醇适量使其溶解,用0.1 mol·L-1盐酸溶液定量制成每1 mL中含帕罗西汀约3.4 μg的溶液,作为对照品溶液。照含量测定项下色谱条件,精密量取上述两种溶液各100 μL,分别注入液相色谱仪,记录色谱图。按外标法以峰面积计算每片的释放量,限度不得过标示量的10%,应符合规定。弃去各溶出杯中酸液,快速用水冲洗后,随即将转篮浸入与酸液相同温度的pH 6.8或pH 7.5缓冲液,转速不变,继续依法操作,经2、4、12 h时,各取溶液适量,滤过,并同时补充相同温度、相同体积的溶出介质,取续滤液作为供试品溶液;另精密称取盐酸帕罗西汀对照品适量,加入甲醇适量使其溶解,用0.05 mol·L-1三羟甲基氨基甲烷缓冲液定量制成每1 mL中含约帕罗西汀25 μg的溶液,作为对照品溶液。照含量测定项下色谱条件,精密量取上述两种溶液各10 μL,分别注入液相色谱仪,记录色谱图。按外标法以峰面积分别计算每片在不同时间的释放量。
2.4 处方筛选与工艺研究
2.4.1 片芯处方筛选
①粘合剂浓度筛选以聚维酮K30作为粘合剂,溶剂为乙醇和水,考察了不同质量分数的聚维酮K30及乙醇对制粒过程和颗粒性状的影响,实验结果见表3
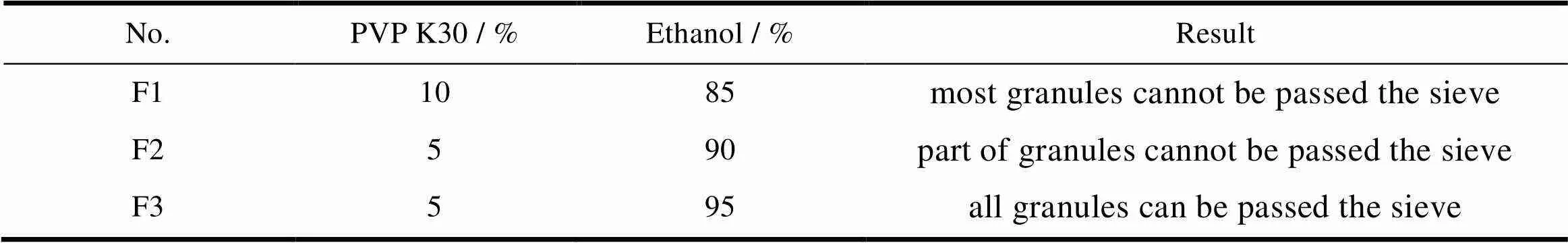
Table 3 The screen experiment of adhesives concentration
结果表明,聚维酮K30的含量越高,乙醇含量越低,制粒结块情况越严重,当聚维酮K30的质量分数为5%、乙醇质量分数为95%时,制粒过程无结块现象。最终确定聚维酮K30和乙醇的质量分数分别为5%和95%是最佳条件。
②阻挡层骨架材料用量筛选:山嵛酸甘油酯为溶蚀性缓释材料、乳糖为水溶性填充剂、羟丙甲纤维素(E5)为亲水凝胶缓释材料,三者比例可调节阻挡层的溶蚀速度从而影响药物的释放速率。控制片芯其他成分不变,调整不同的山嵛酸甘油酯/乳糖/HPMC(E5)比例,具体处方见表4,pH 7.5介质释放度曲线见图1。

Table 4 The screen experiment of polymers amount in barrier-layer

Fig. 1 The effect of polymers amount of barrier-layer on drug release
图1 阻挡层骨架材料对释放度的影响
结果表明,山嵛酸甘油酯用量在40%时,释放速度明显减慢;山嵛酸甘油酯用量在30%时,随乳糖用量减少,羟丙甲纤维素(E5)用量增加,释放速度呈减慢趋势。
③含药层骨架材料用量筛选:羟丙甲纤维素(K4M)作为含药层缓释骨架材料、乳糖作为含药层填充剂,分别考察羟丙甲纤维素(K4M)和乳糖的不同用量对释放度的影响,具体处方见表5,pH 7.5介质释放度曲线见图2。

Table 5 The screen experiment of polymers amounts in drug-layer

Fig. 2 The effect of polymers amounts of drug - layer on drug release
图2 含药层骨架材料对释放度的影响
结果表明,随着羟丙甲纤维素(K4M)用量增加、乳糖用量减少,释放速度呈减慢趋势。
2.4.2 肠溶包衣处方影响因素考察
①抗粘剂用量对释放度的影响 选择甲基丙烯酸-丙烯酸乙酯共聚物水分散体(尤特齐L 30 D-55)作为肠溶包衣材料,包衣处方需加入滑石粉作为抗粘剂,枸橼酸三乙酯作为增塑剂,滑石粉用量过多可能会影响到肠溶衣膜的致密性,从而影响肠溶衣的耐酸性,同时影响药物的释放度。固定枸橼酸三乙酯用量为聚合物的15%,分别考察滑石粉的用量为聚合物的35%、50%的处方在0.1 mol·L-1盐酸及pH 7.5介质中释放度的变化,具体处方见表6,耐酸性结果见表7,释放度结果见图3。

Table 6 The screen experiment of talc in enteric coating

Table 7 The effect of talc amounts on release in 0.1 mol·L-1 HCL medium
结果表明,滑石粉用量在35%~50%之间对耐酸性无影响,随用量增加,释放速度呈加快趋势,本文选择相当于35%聚合物的加入量。
②包衣增重对释放度的影响包衣处方如下:36.628 g尤特齐L 30 D-55、3.848 g滑石粉、1.646 g枸橼酸三乙酯、67.762 g纯化水。
配制方法:称取处方量的滑石粉、枸橼酸三乙酯分散到纯化水中,再加入处方量尤特齐L 30 D-55搅拌均匀。
包衣参数:进风温度为40~60 ℃,蠕动泵转速为7~17 r·min-1,雾化压力为0.1~0.2 MPa,片床温度为30~50 ℃。
分别考察包衣增重为6%、8%、11%时,对药物释放行为的影响,耐酸性结果见表8,pH 6.8介质释放度曲线见图4。

Table 8 The effect of coating weight on release in 0.1 mol·L-1 HCL medium
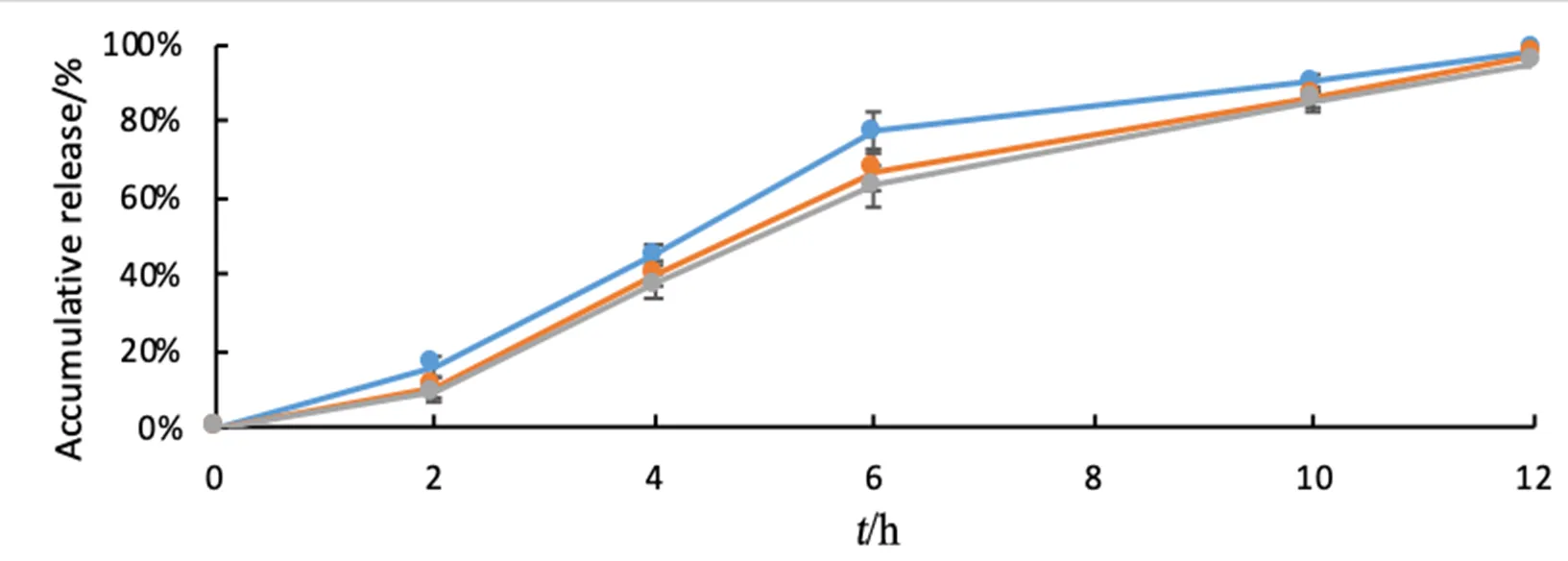
—coating weight 6%;—coating weight 8%;—coating weight 10%
结果表明,以尤特齐L 30 D-55作为肠溶包衣材料,包衣增重6%~10%,均可有效地耐酸,随包衣增重增加,释放速度减慢,本文选择包衣增重8%~10%作为包衣增重的控制范围。
2.4.3 片芯硬度影响因素考察
按2.4.4项下处方制备双层片,分别考察片芯平均硬度为6、5、4 kgf时,对药物释放度的影响,pH 6.8介质释放度曲线见图5。结果表明片芯硬度增加,对药物释放速度无显著影响。
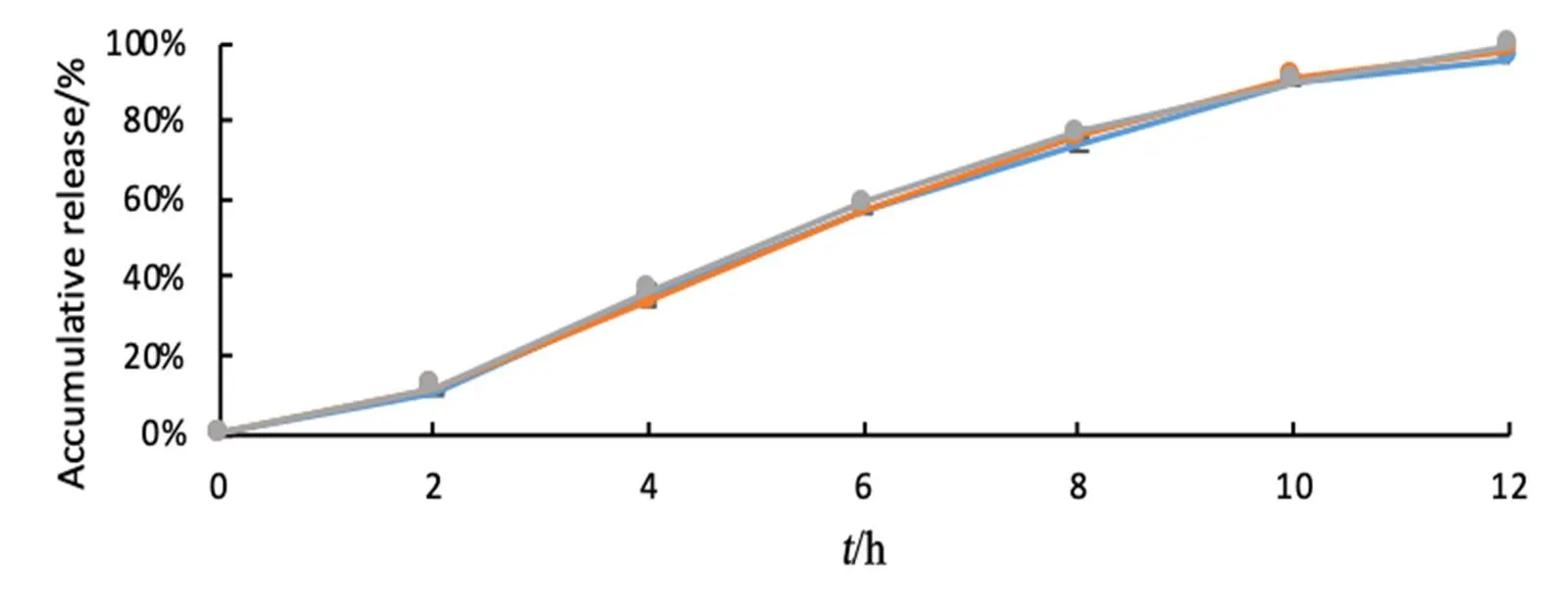
—hardness 6 kgf;—hardness 5 kgf;—hardness 4 kgf
2.4.4 药物动力学试验样品的制备及释放度考察
按2.2.1及2.2.2项下制备方法制备药物动力学试验样品,处方见表9。

Table 9 The composition of pharmacokinetic test samples
按上述处方制备盐酸帕罗西汀肠溶缓释片,按照“释放度测定方法”项下方法分别测定其在0.1 mol·L-1盐酸、pH 6.8和pH 7.5缓冲液中的释放度,以f因子方法和参比制剂Paxil CR比较相似性,样品在0.1 mol·L-1盐酸介质均无溶出,在pH 6.8和pH 7.5介质释放度曲线见图6、7。


Fig. 6 The drug release of pharmacokinetic test samples in pH 7.5 medium
图6 药物动力学试验样品释放度曲线-pH7.5介质
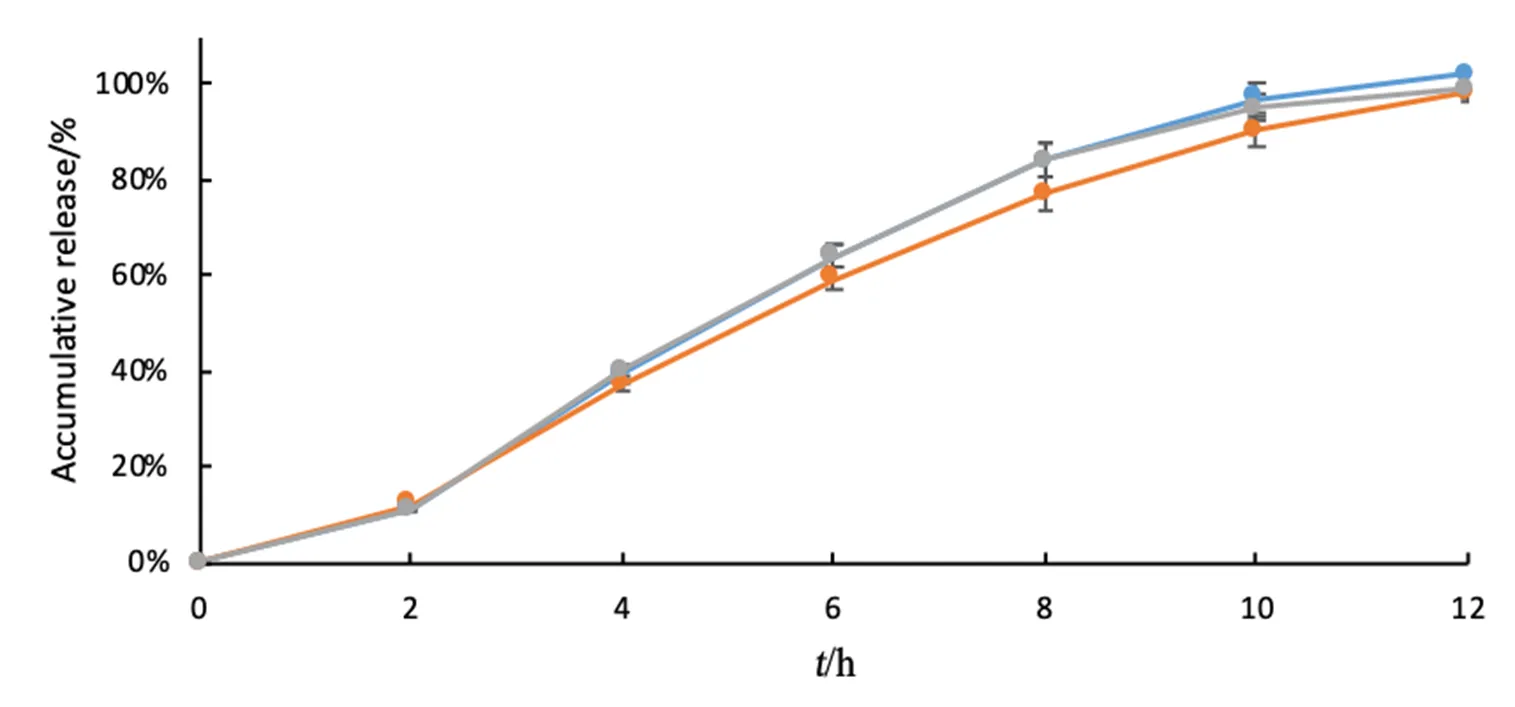
—Paxil CR;—T1;—T2
以PAXIL CR为参比制剂,计算释放曲线的相似因子f,结果见表10。

Table 10 The similar factor f2 of test preparation and reference preparation
结果表明,两个处方在pH 6.8和pH 7.5介质中自制制剂和参比制剂的相似因子f均大于50,释放曲线相似。
2.5. 药物动力学实验
采用随机、开放、三制剂、三周期交叉试验设计,14名健康成年志愿者在进食状态下单次口服2批受试制剂(盐酸帕罗西汀肠溶缓释片,25 mg)与1批参比制剂(PAXIL CR,25 mg),测定血药浓度。药物动力学参数结果见表11,血药浓度-时间曲线见图8。
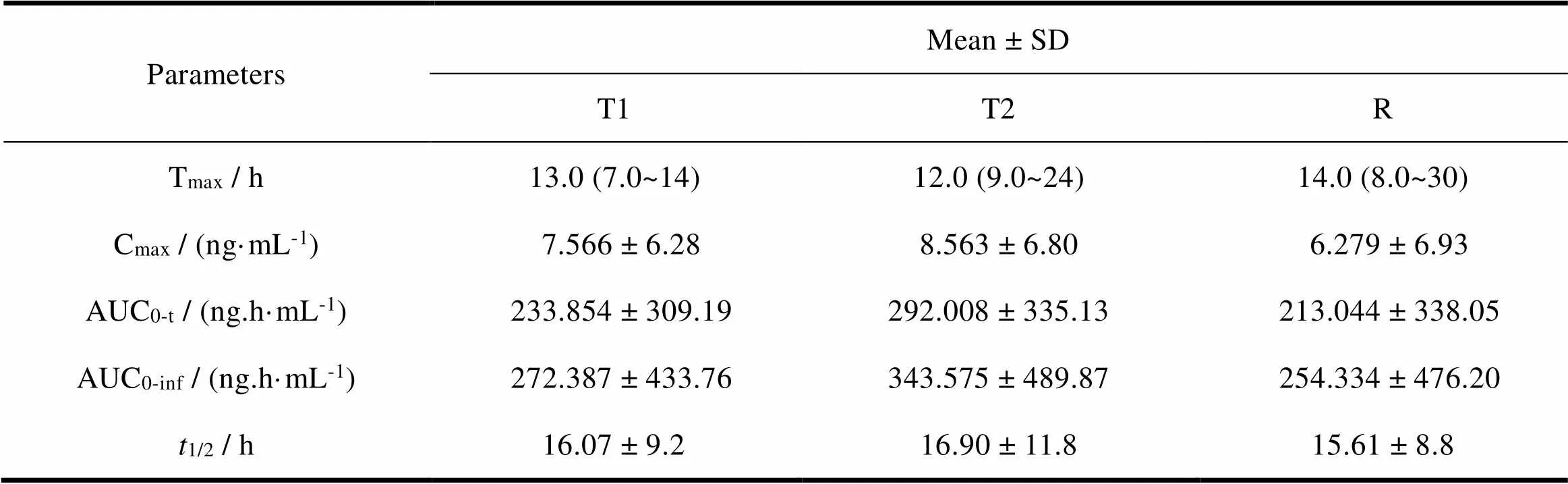
Table 11 The result of pharmacokinetic parameters (n=14)

—T1;—T2;—R
与药物暴露相关的主要药动学参数Cmax和AUC经对数转换后以多因素方差分析(ANOVA)进行显著性检验,然后用双单侧检验、计算90%置信区间评价等效性。帕罗西汀Cmax、AUC0-t和AUC0-∞经对数转换的平均生物等效性分析结果如表12。
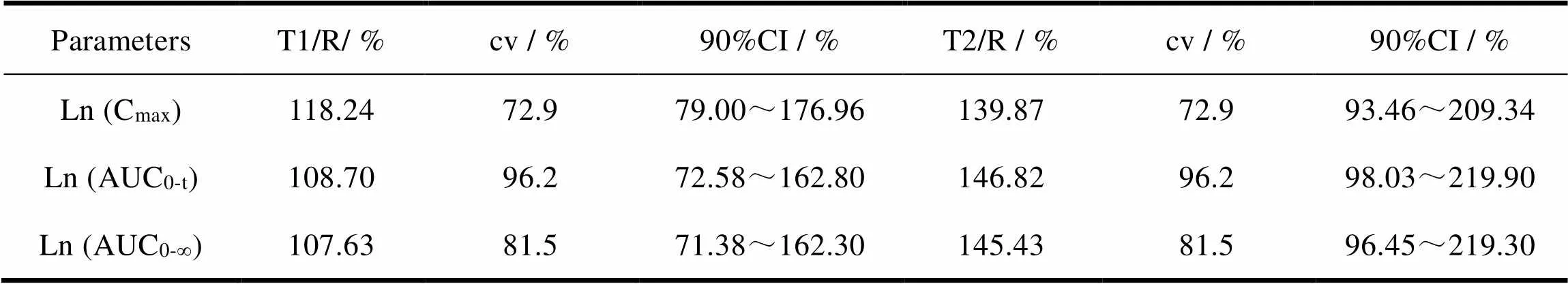
Table 12 The statistical results of Pharmacokinetic parameters
结果表明R约14 h达到峰值,T1约13 h达到峰值、T2约12 h达到峰值,提示T1、T2吸收比R快。T1的Ln(Cmax)与R比值的均值为118.24%,ln(AUC0-t)与R比值的均值为108.70%,显示受试制剂相较参比制剂其整体吸收量稍偏高且吸收速度偏快;T2的Ln(Cmax)与R比值的均值为139.87%,Ln(AUC0-t)与R比值的均值为146.82%,显示受试制剂相较与参比制剂其整体吸收量显著偏高且吸收速度显著偏快。
3 讨论
a. 本文采用了HPLC法用于帕罗西汀含量测定和释放度测定,经方法学验证,所建立的各分析方法专属性强、重现性好、准确度高。简便、快速,可作为质量控制和稳定性研究的检测方法。
b. 本文采用湿法制粒工艺分别制备两层颗粒,压制双层片,并包肠溶衣。片芯的一层是以山俞酸甘油酯为主的溶蚀屏障层,另一层为以羟丙甲纤维素(K4M)含药亲水骨架层。肠溶包衣延迟药物从体内开始释放的时间,待缓释片进入肠道后,肠溶包衣溶解,屏障层开始溶蚀,含药骨架层吸水形成凝胶,药物通过扩散从亲水凝胶层释放,溶蚀屏障层的作用是抑制含药骨架层与其接触面的释放速度。以释放度为主要评价指标,考察了不同处方用量及工艺参数对释放度的影响,屏障层中主要影响释药行为的辅料是山俞酸甘油酯和羟丙甲纤维素(E5),随山俞酸甘油酯和羟丙甲纤维素(E5)用量增加,药物释放速度减慢;含药层中主要影响释药行为的辅料是羟丙甲纤维素(K4M),随羟丙甲纤维素(K4M)用量增加,药物释放速度减慢;肠溶包衣处方中滑石粉用量会影响释药速度,由于滑石粉可以减少包衣膜的致密性,随滑石粉用量增加,药物释放速度加快;压片硬度对药物释放速度影响不显著;肠溶包衣增重会影响释药速度,随包衣增重增加,药物释放速度减慢。
c. 本文通过调节处方中羟丙甲纤维素(K4M)和山俞酸甘油酯用量,得到两个不同释放速度的自制制剂进行人体药物动力学实验,其中T1处方羟丙甲纤维素(K4M)含量高于T2处方,山俞酸甘油酯含量低于T2处方。药物动力学实验结果显示,自制制剂T1释放速度慢于自制制剂T2,体内吸收速率慢于T2制剂,吸收程度低于T2制剂,说明对体外释放及体内吸收影响较大的辅料为羟丙甲纤维素(K4M)。
d. 本文将两批不同释放速度的自制制剂T1、T2和参比制剂R进行药物动力学试验,对药动学参数进行对比及分析。对各药动学参数进行分析,受试制剂T1 的相对生物利用度为108.70%,显示受试制剂相较于参比制剂其整体吸收量稍偏高,由于CV大于70%,个体变异性大,试验样本量太少,90%置信区间未达到生物等效要求,如果要达到生物等效的结论,需要将受试者例数放大,可依此试验方案进一步扩大样本量开展正式试验。受试制剂T2的相对生物利用度为146.82%,显示受试制剂相较参比制剂其整体吸收量显著偏高且吸收程度明显偏快,即使进一步扩大样本量,也无法达到生物等效。
e. 本文开发的溶出曲线方法可以反映自制制剂处方、工艺等因素变化对药物体外释放行为的影响,受试制剂T2释放速度快于受试制剂T1,其整体吸收速率显著快于受试制剂T1,吸收程度显著高于受试制剂T1,显示药物体外释放速度和体内吸收具有一定相关性。
f. T1、T2受试制剂和参比制剂溶出曲线相似性用2相似因子法评价,2均大于50,人体药动学试验结果显示,T2与参比制剂无法生物等效,说明2相似因子法可用于处方筛选,但不能完全预测盐酸帕罗西汀肠溶缓释片的体内等效性,且该药物的体内吸收个体变异性大,研究者最终应对开发的制剂进行生物等效性预试验,为正式试验的样本量提供依据及预测正式试验的成功率。
[1] 孟新玲, 房江山, 刘远新, 等. 盐酸帕罗西汀片对帕金森病患者合并抑郁和焦虑症状的临床疗效[J]. 中国临床药理学杂志, 2013,29(6): 403-405.
[2] 赵海霞, 张翼萍, 马建英, 等. 盐酸帕罗西汀对卒中后抑郁及神经功能恢复的影响[J]. 武警医学院学报, 2010, 19(2): 111-113.
[3] FDA.PAXIL CR® (paroxetine hydrochloride) Controlled-Release tablets prescribing information[EB/OL]. [2016- 12-16]. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020936s049lbl.pdf.
[4] PMDA.パキシルCR錠12.5mg/パキシルCR錠25mg[EB/OL]. [2017-04-18]. https://www.info.pmda.go.jp/go/pack/1179041G1020_1_12/?view=frame&style=XML&lang=ja.
[5] 谢向阳, 任宏英. 盐酸帕罗西汀肠溶缓释片的制备及体外释放研究[J]. 西北药学杂志, 2016,31(4): 395-399.
[6] 刘海洲, 张媛媛, 刘均洪. 盐酸帕罗西汀控释微丸的制备及质量评价[J]. 抗感染药学, 2009,6(4): 241-244.
[7] 尹辉. 盐酸帕罗西汀肠溶缓释片的制备及体外释放研究[J]. 药学杂志, 2016,28(11): 19-21.
[8] 胡献跃, 郑一美. 盐酸帕罗西汀缓释片的研制[J]. 中国药业, 2016,25(17): 31-35.
[9] 史密丝克莱恩比彻姆有限公司. 帕罗西汀控释组合物:中国, 96196819.2[P]. 1996-07-19.
[10] 美国药典委员会. 美国药典40版[M]. 美国药典委员会, 2017: 5583-5586.
[11] 国家药典委员会. 中国人民共和国药典:2015年版四部[M]. 北京:中国医药科技出版社, 2015: 121-124.
Preparation and evaluationrelease andpharma-cokinetic of paroxetine hydrochloride enteric-coated sustained-release tablets
CAI Yanxia, CHENG Gang*
(110016)
To prepare enteric-coated sustained-release tablets of paroxetine hydrochloride, and evaluate itsrelease and pharmacokinetic parameters.Hypromellose and glyceryl behenate were used to prepare sustained release tablets by application of double compression and enteric coating technology.release was detected by HPLC, and the release behavior similarity was investigated by comparison between the reference and self-prepared tablets. Similar factor2was used to evaluate the similarity of release curves. A single-dose tablets of reference and two batches of self-made preparations with different formulations were given to 14 healthy volunteers, and pharmacokinetic parameters were estimated.Therelease data of the two self-made preparations and the reference showed that the2was more than 50, and the relative bioavailability was 108.70% and 146.82%, respectively.The formulated paroxetine hydrochloride enteric-coated sustained-release tablets exhibit sustained release characteristics. The result suggests that one of the self-made preparations will be bioequivalent with the reference if increase the test sample, while the other self-made preparation cannot be bioequivalent. There existed correlation between therelease andabsorption of paroxetine hydrochloride enteric sustained-release tablets, but the2can not be used to evaluate the bioequivalence.
paroxetine hydrochlorideenteric-coated sustained release tablets;release; pharmaco-kinetic; similar factorf
2019-08-22
蔡燕霞(1977-), 女(汉族), 广东湛江人, 硕士研究生, E-mail caiyanxia@salubris.com;
程刚(1963-), 男(汉族), 辽宁康平人, 教授, 博士, 博士生导师, 主要从事药剂学的研究, Tel. 024-23986326,E-mailchenggang63@ Hotmail.com。
R943
A
(2020)03–0153–12
10.14146/j.cnki.cjp.2020.03.003
