二黄益肾汤质量标准研究
2020-06-17田雪芬郭文厂李冬冬黄秀贞
田雪芬,郭文厂,李冬冬 ,黄秀贞
(山东省济宁市中医院,山东 济宁 272100)
二黄益肾汤系山东省济宁市中医院肾病科的临床经验方,由丹参、炒白术、大黄、炙淫羊藿等10味中药组方,具有补益脾肾、降浊排毒、活血通络之功效,用于气虚湿瘀型慢性肾功能不全(CKD 3~4期)的治疗。为确定其制备工艺的合理性,全面有效控制质量,保障临床用药安全、有效,本研究中采用薄层色谱(TLC)法对方中大黄和白术进行了定性鉴别,采用反相高效液相色谱(RP-HPLC)法测定丹参中丹参素和原儿茶醛及淫羊藿中淫羊藿苷的含量,为进一步完善二黄益肾汤的质量标准提供依据。现报道如下。
1 仪器与试药
1.1 仪器
LC-20AT型高效液相色谱仪(日本岛津公司);AB-135S型电子天平(Mettler Toledo公司,精度为十万分之一);KQ-500DB型超声波清洗器(昆山市超声仪器有限公司,功率为 300 W,频率为 25 kHz);ZF-8型暗箱三用紫外分析仪(上海一科仪器有限公司)。
1.2 试药
大黄对照药材(批号为 120984-201202),白术对照药材(批号为120925-201611),丹参素钠对照品(纯度≥98%,批号为110855-201614),原儿茶醛对照品(纯度≥99%,批号为110810-201608),淫羊藿苷对照品(纯度≥98%,批号为110766-201721),均购自中国食品药品检定研究院;二黄益肾汤(济宁市中医院制剂室自制,批号分别为 20181217,20181219,20181221,规格为每瓶 150 mL);甲醇,乙腈(色谱纯,Avantor Performance Materials,Inc.,USA;GC≥99.9% );娃哈哈纯净水;丹参(批号为201803201)、炒白术(批号为201804122)、大黄(批号为 201801291)、炙淫羊藿(批号为201803266)等10味药材均由济宁邦尔中药饮片有限公司提供,经济宁市中医院刘会芳副主任中药师鉴定为正品,均符合《中国药典(一部)》相关规定。
2 方法与结果
2.1 样品制备
按处方称取丹参、炒白术、大黄、炙淫羊藿等10味药材,其中炒白术、砂仁、莪术混合加10倍量水,浸泡0.5 h,采用水蒸气蒸馏法提取挥发油,冷藏备用;大黄加 12倍量水,浸泡 0.5 h,微沸 5 min,滤过,备用;其余6味药材与提取挥发油的药渣和蒸馏后的水溶液、大黄药渣混合,加 14倍量水,浸泡 0.5 h,煎煮 1 h,倾出煎液;药渣再加10倍量水,煎煮40 min,倾出煎液,合并2次煎液,滤过;将滤液浓缩至约350 mL,放冷至室温,搅拌下加入大黄滤液,冷藏、静置过夜,滤过,滤液加热至沸,放冷,加入上述挥发油,加纯化水至450 mL,混匀,即得样品。
2.2 薄层色谱鉴别
大黄:取样品(批号为 20181014)30 mL,加盐酸 2 mL,加热回流 30 min,立即冷却,用乙醚提取 2次,每次40 mL,合并乙醚液,溶剂挥干,残渣加三氯甲烷1 mL使溶解,作为供试品溶液。取大黄对照药材0.5 g,加甲醇20 mL,超声处理20 min,滤过,滤液蒸干,残渣加水10 mL使溶解,按供试品溶液制备方法制备对照药材溶液。取除大黄外的其余药材,按处方和样品制备工艺制备不含大黄的阴性对照品,按供试品溶液制备方法制备阴性对照药材溶液。照2015年版《中国药典(一部)》通则0502中 TLC法,吸取供试品溶液8 μL和对照药材溶液6 μL,分别点于同一硅胶G薄层板上,以石油醚(30~60℃)-甲酸乙酯 -甲酸(7.5∶2.5∶0.6,V/V/V)的上层溶液为展开剂,展开,取出,晾干,置氨蒸气中熏至斑点显色清晰[1-3]。供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,置紫外光灯(365 nm)下检视,显相同颜色的荧光斑点,阴性对照无干扰。详见图1。
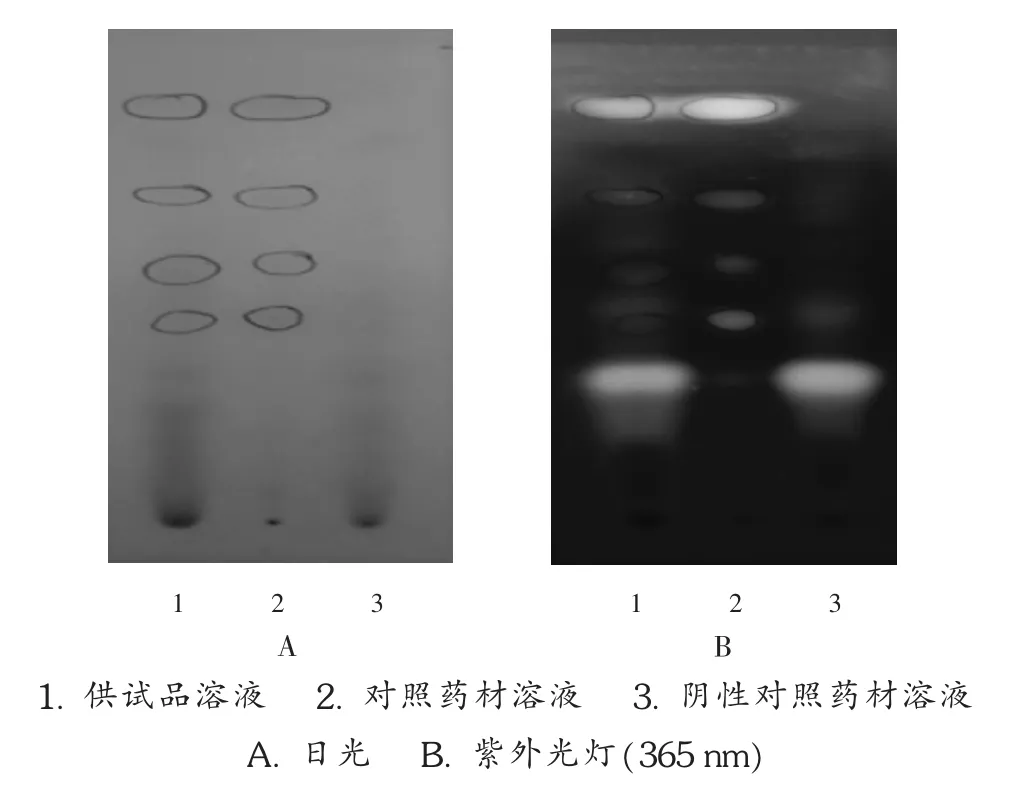
图1 大黄薄层色谱图
白术:取样品(批号为 20181014)90 mL,加乙醚振摇提取2次,每次100 mL,合并乙醚液,溶剂挥干,残渣加环己烷1 mL使溶解,作为供试品溶液。另取白术对照药材 2.0 g,加水 50 mL,加热回流 30 min,放冷,滤过,用乙醚提取2次,每次60 mL,合并乙醚液,挥干,残渣加环己烷1 mL使溶解,作为对照药材溶液。取除白术外的其余药材,按处方和样品制备工艺制备不含白术的阴性对照品,按供试品溶液制备方法制备阴性对照药材溶液。照 2015年版《中国药典(一部)》通则0502中 TLC法,吸取上述3种溶液各10 μL,分别点于同一硅胶G薄层板上,以石油醚(60~90℃)-乙酸乙酯(2∶1,V/V)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,在105℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视[1,4]。供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照无干扰。详见图2。

图2 白术薄层色谱图
2.3 丹参素和原儿茶醛含量测定
2.3.1 色谱条件[5-7]
色谱柱:Purospher star RP-C18柱(250 mm ×4.6 mm,5 μm);流动相:乙腈(流动相 A),0.4% 冰醋酸 - 水溶液(流动相 B),0~35 min 时 3% ~10% (A);检测波长:280 nm;柱温:30 ℃;流速:0.8 mL /min;进样量:8 μL。
2.3.2 溶液制备
分别取丹参素钠对照品1.07 mg和原儿茶醛对照品0.27 mg,精密称定,置10 mL容量瓶中,加甲醇至刻度,摇匀,即得质量浓度为0.097 6 g/L的丹参素对照品贮备液(1 mg丹参素钠相当于0.912 mg丹参素)和0.027 0 g/L的原儿茶醛对照品贮备液。按2.1项下方法制备样品,精密量取5 mL,置10 mL棕色容量瓶中,加50%甲醇稀释至刻度,摇匀,离心,取上清液,过滤,即得供试品溶液。
2.3.3 方法学考察
专属性试验:按处方和制备工艺制备不含丹参的阴性对照样品,按供试品溶液制备方法制备阴性对照品溶液。分别精密吸取上述3种溶液各10 μL,注入液相色谱仪,按拟订色谱条件进样测定。色谱图见图3。
线性关系考察:精密量取上述对照品贮备液,用甲醇逐级稀释成 6.1,12.2,24.4,48.8,73.2,97.6 μg /mL系列质量浓度的丹参素对照品溶液和 1.688,3.375,6.750,13.500,20.250,27.000 μg /mL 系列质量浓度的原儿茶醛对照品溶液,精密吸取上述对照品溶液各16μL,按拟订色谱条件依法分别进样,记录峰面积。以质量浓度(X)为横坐标、峰面积(Y)为纵坐标进行线性拟合,得丹参素的回归方程 Y丹=13 035.912 X丹-4 369.223,r=1.000 0(n=6);原儿茶醛的回归方程 Y原=71 310.237 X原-6 841.460,r=1.000 0。结果表明,丹参素和原儿茶醛的质量浓度分别在 6.1 ~ 97.6 μg/mL,1.688 ~27.000 μg/mL 范围内与峰面积线性关系良好。

图3 丹参高效液相色谱图
精密度试验:精密吸取上述对照品混合贮备液8 μL,按拟订色谱条件进样测定,连续进样6次。结果丹参素和原儿茶醛峰面积的 RSD分别为 0.96%和 1.40%(n=6),表明仪器精密度良好。
稳定性试验:取样品(批号为20181219),精密吸取供试品溶液 10 μL,按拟订色谱条件分别于 0,4,8,12,16,24 h时进样测定。结果丹参素和原儿茶醛含量的RSD 分别为 2.67%和 2.08% (n=6),表明供试品溶液在24 h内稳定性良好。
重复性试验:取样品(批号为20181219),依法制备供试品溶液6份,按拟订色谱条件进样测定,各进样1次。结果丹参素和原儿茶醛含量的 RSD分别为2.54%和1.21%(n=6),表明方法重复性良好。
加样回收试验:平行取6份已知含量的样品(批号为20181219)45 mL,分为平行2份3个水平,分别加入含量相当量80%,100%,120%的丹参素和原儿茶醛混合对照品,依法制备供试品溶液,进样10 μL,记录色谱图,测量丹参素和原儿茶醛的峰面积,代入回归方程计算。结果丹参素的平均回收率为 99.58%,RSD为2.36% (n= 6);原儿茶醛的平均回收率为 101.44% ,RSD 为 1.30%(n=6)。
定量限、检测限确定:取上述对照品贮备液逐级稀释,取信噪比(S/N)为10的作为定量限,丹参素和原儿茶醛的定量限分别为 11.146,1.013 ng;取 S /N 为 3的作为检测限,丹参素和原儿茶醛的检测限分别为1.793 ng 和 0.006 80 ng。
2.3.4 样品含量测定
取3批样品,按供试品溶液制备方法平行制备2份,按拟订色谱条件分别进样测定,计算样品中丹参素和原儿茶醛的含量。结果见表1。

表1 样品含量测定结果(mg/100 mL,n=3)
2.4 淫羊藿苷含量测定
2.4.1 色谱条件[8-9]
色谱柱:Purospher Star RP-C18柱(250 mm×4.6 mm,5 μm);流动相:以乙腈 -0.1%磷酸溶液(26 ∶74,V/V);流速:0.8 mL /min;检测波长:269 nm;进样量:8 μL。
2.4.2 溶液制备
取淫羊藿苷对照品 1.29 mg,精密称定,置 10 mL容量瓶中,加甲醇至刻度,摇匀,即得质量浓度为0.129 g/L的淫羊藿苷对照品贮备液。精密吸取样品(批号为20181217)5 mL,置50 mL容量瓶中,加甲醇40 mL,超声处理(功率为 300 W,频率 25 kHz) 20 min,放冷,加甲醇至刻度,摇匀,滤过,续滤液浓缩至5 mL,即得供试品溶液。
2.4.3 方法学考察
专属性试验:按处方和制备工艺制备不含淫羊藿的阴性对照样品,按供试品溶液的制备方法制成阴性对照品溶液。分别精密吸取上述3种溶液各10 μL,注入液相色谱仪,按拟订色谱条件进样测定。色谱图见图4。
线性关系考察:精密量取上述对照品贮备液,用甲醇逐级稀释成 8.063,16.125,32.250,64.500,96.750,129.000 μg/mL系列质量浓度的对照品溶液,精密吸取上述对照品溶液各16 μL,按拟订色谱条件及方法分别进样,记录峰面积。以对照品溶液质量浓度(X)为横坐标、峰面积(Y)为纵坐标进行线性拟合,得淫羊藿苷的回 归 方 程 Y=33 267.217X+10 163.538, r=1.000 0(n=6)。结果表明,淫羊藿苷质量浓度在 8.063~129.000 μg /mL 范围内线性关系良好。
精密度试验:精密吸取上述对照品贮备液10 μL,按拟订色谱条件连续进样6次,记录淫羊藿苷的峰面积。结果的 RSD为0.48%(n=6),表明仪器精密度良好。
稳定性试验:取样品(批号为20181217),依法制备供试品溶液,精密吸取5 μL,按拟订色谱条件分别于0,4,8,12,16,24 h 时进样测定。结果淫羊藿苷含量的RSD为2.49%(n=6),表明供试品溶液在24 h内稳定性良好。
重复性试验:取样品(批号为20181217),依法制备供试品溶液6份,按拟订色谱条件进样测定。结果淫羊藿苷含量的 RSD为 1.84%(n=6),表明方法重复性良好。
加样回收试验:平行取6份已知含量的样品各10mL,分为平行2份3个水平,分别加入含量相当量80%,100%,120%的淫羊藿苷对照品粉末,依法制备供试品溶液,每次进样5 μL,记录色谱图,测量淫羊藿苷的峰面积,代入回归方程计算。结果淫羊藿苷的平均回收率为 99.80%,RSD 为 1.08%(n=6)。
定量限、检测限确定:取上述对照品贮备液逐级稀释,取信噪比(S/N)为10的作为定量限,淫羊藿苷的定量限为 1.954 ng;取 S/N为 3的作为检测限,淫羊藿苷的检测限为0.279 ng。
2.4.4 样品含量测定
取3批(批号分别为20181217,20181219,20181221)样品,依法平行制备2份供试品溶液,按拟订色谱条件分别进样测定,计算含量。结果见表1。
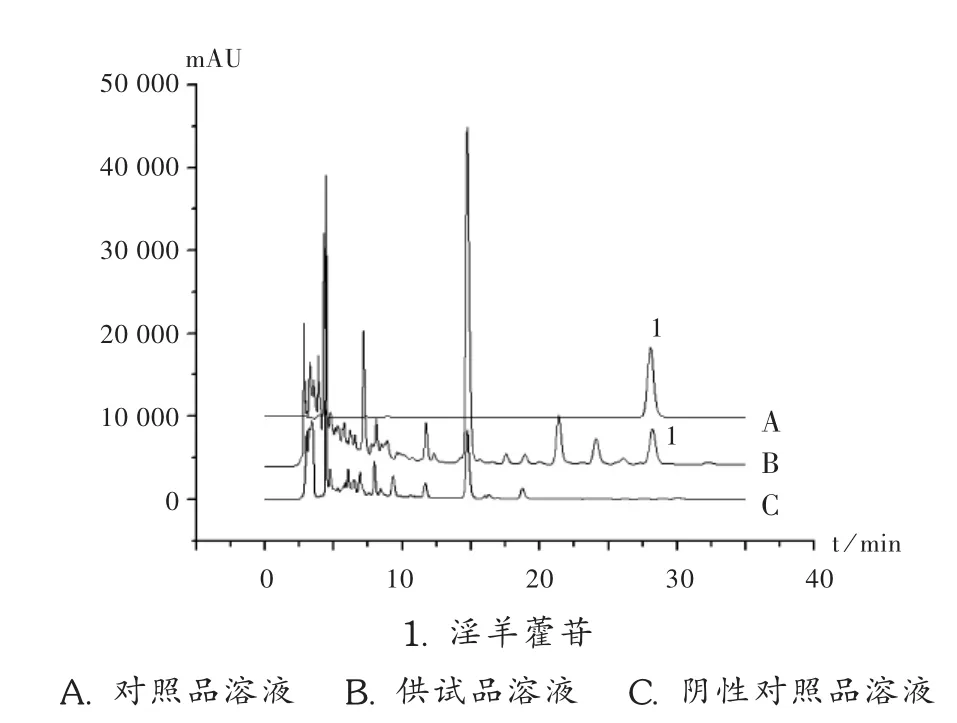
图4 淫羊藿高效液相色谱图
3 讨论
3.1 薄层色谱鉴别要求
对白术进行薄层色谱鉴别时,105℃加热至紫外灯下检视,主斑点不清晰,105℃加热时间略长,但羧甲基纤维素未碳化变黑后,主斑点显色清晰。对大黄进行薄层鉴别时,参考2015年版《中国药典(一部)》中八正合剂相应药材的鉴别方法,方法简单、合理,斑点显色清晰,分离效果好。
3.2 丹参中有效成分含量分析指标确定
丹参中有效成分主要为脂溶性的丹参酮类和水溶性的酚酸类化合物,2015年版《中国药典(一部)》规定了丹参的含量测定成分为丹参酮类和丹酚酸B。丹参酮类化合物具有脂溶性高、对光和温度不稳定、半衰期短等缺点。GONG等[10]报道了丹酚酸B受热后降解为丹酚酸A、咖啡酸、丹酚酸C、丹参素、原儿茶醛等,其降解产物可作为以丹酚酸B为指标进行含量分析的物质基础。原儿茶醛具有抗氧化、抑制血小板聚集、改善肾脏血循环、减轻急性肾损伤引起的肾小管坏死的作用[11];丹参素可降低肾小管上皮细胞转分化为成纤维细胞的比率,维持肾小管上皮细胞的正常形态[12]。对含有丹参的中药复方制剂进行含量测定时,多以丹参素和原儿茶醛作为指标[13]。故本试验中选择了丹参中的2种水溶性成分(丹参素和原儿茶醛)为指标进行含量分析。
3.3 流动相选择
流动相采用了甲醇(B)-0.4%冰醋酸水溶液(C)等度洗脱[14],结果发现峰形拖尾较严重,样品中化合物的峰高被压得很低。选择流动相乙腈(B)-0.4%冰醋酸水溶液(C)梯度洗脱,0~7 min时 2% ~5%(B),7~35 min 时5% ~13%(B)[5],但原儿茶醛与其前面一个未知峰未分开。经多次调整梯度洗脱的流动相比例,但峰形越来越无法分开,因此减少梯度洗脱比例变化的幅度,采用乙腈(B)-0.4% 冰醋酸水溶液(C)梯度洗脱,0~40 min时 3% ~9% (B)、0~35 min 时 3% ~11% (B),前者出峰时间较长,后者原儿茶醛与其前面一个峰相邻太近。最后采用0~35 min时3% ~10%(B)的比例,则丹参素和原儿茶醛可完全与杂峰分离,且峰形和出峰时间较好。
3.4 淫羊藿流动相和检测波长选择
淫羊藿中多以淫羊藿苷为指标进行含量测定[15-16]。曾考察乙腈 -水(25∶75,26∶74,28∶72,30∶70,V/V),其中流动相比例为25∶75时出峰时间太长,为28∶72和30∶70时基线不稳,为26∶74时样品分离度良好,略有拖尾。故确定用乙腈 -0.1% 磷酸水溶液(26∶74,V/V)作为淫羊藿苷的流动相。使用二极管阵列检测器(PDA)进行了全波长扫描(190~800 nm),结果淫羊藿样品在269 nm波长处淫羊藿苷有较大吸收,且杂质成分干扰少,故选择269 nm作为淫羊藿苷的检测波长。
