土壤中砷的来源及迁移释放影响因素研究进展①
2020-06-15安礼航刘敏超张建强陈志良
安礼航,刘敏超,张建强,黄 玲,陈志良*
(1 五邑大学生物科技与大健康学院,广东江门 529020;2 生态环境部华南环境科学研究所,广州 510655;3 广东省农田重金属污染土壤治理与修复工程技术研究中心,广州 510655)
砷是具有急性和慢性毒性有毒准金属元素,被国际癌症研究机构(IARC)定义为I 类致癌物质。人体砷中毒会导致肺损伤、外周神经损伤、皮肤病或心血管病等症状,严重则致瘫痪和致癌致畸[1]。土壤中的砷含量和饮用水中的砷浓度具有密切联系,1993 年世界卫生组织(WHO)建议将饮用水中砷含量标准由50 μg/L降低为10 μg/L,中国、印度以及美国等国家部分水源中砷含量均超过该标准[2]。全世界约有2 亿人的饮用水安全受到高砷暴露的威胁[3],尤其是东南亚国家,中国约有1 958 万人生活在地下水砷超标的高风险地区。
我国砷污染问题尤为突出,地下水含砷量在10 μg/L以上的地区总面积达5.8×105km2,土壤中砷平均含量为11.2 mg/kg,约为世界平均值(7.2 mg/kg)的1.5倍[4]。土壤中砷来源于自然本底与人类活动,特别是由于人类活动,如矿山开采、冶炼、施肥、农药等导致大量的砷进入土壤环境。2014 年全国土壤污染状况调查公报显示,全国土壤总的超标率为16.1%,镉、汞、砷、铜、铅5 种无机污染物点位超标率分别为7.0%、1.6%、2.7%、2.1%、1.5%,砷为全国土壤重金属污染物中的首要污染物[4]。水稻是我国受砷污染的主要粮食作物,我国人均砷摄取量约为42 µg/d,远高于西方国家,相当于美国、加拿大、澳大利亚及法国总砷摄入量的4 倍 ~ 4.7 倍,其中通过大米摄入的砷占总砷摄入量的60%[5],较高含砷量的稻米使得以之为主食的人民的健康受到严重威胁。近年来许多学者致力于砷的地球化学行为和降低砷对人体毒害的研究,已取得了一系列的成果,但砷的环境行为受诸多因素的影响,因素间的相互作用机理复杂,目前对这方面鲜有综合性分析。本文综述了环境中砷的来源及影响砷迁移转化的主要因素,为解决土壤砷污染问题提供依据。
1 砷的主要来源
1.1 人为来源
1.1.1 工业采矿和排废 矿物资源开发和工业废物排放(如冶炼和化石燃料燃烧产物)是土壤砷的主要人为来源。常见的矿物资源有含砷黄铁矿增生体(Fe(S,As)2)、毒砂(FeAsS)、斜方砷铁矿(FeAs2)、雄黄(AsS)、雌黄(As2S3),辉砷钴矿(CoAsS)、红砷镍矿(NiAs)和臭葱石(FeAsO4·2H2O)[6],其中含砷量相对较高的是含砷黄铁矿增生体,其砷含量超过 100 g/kg(最高达190 g/kg)[7],含砷量最高的是硫化物和氧化物矿物(铁矿)(表1),其他常见的硫酸盐成岩矿物(碳酸盐和硅酸盐)、硅酸盐矿物(包括石英、长石、云母、闪石)和碳酸盐矿物中的砷含量往往较低(一般小于5 mg/kg)。有色金属冶炼过程排废是最重要的砷污染源之一,全球该行业每年排放砷近10 万t,其中90% 左右的砷来自铜、铅、锌等有色金属冶炼行业(图1)[9]。
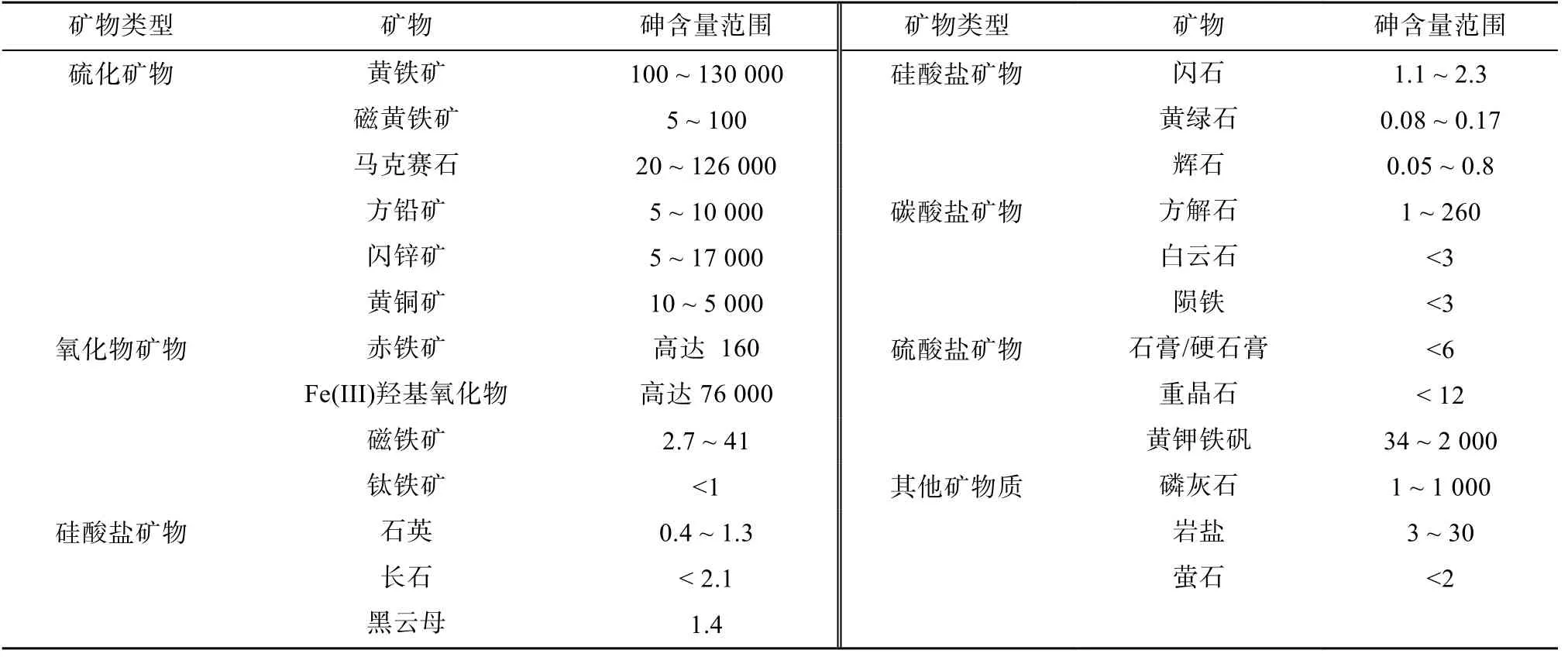
表1 矿物中砷的典型含量范围[6-8] (mg/kg)Table 1 Typical ranges of As concentrations in minerals

图1 全球冶金工业砷年排放量Fig. 1 As emissions from the global metallurgical industry
1.1.2 农业排放 农业生产过程中使用农药和磷肥是土壤砷的另一个重要来源。牲畜养殖中常用多种含砷化合物如洛克沙胂等作为饲料添加剂,该类添加剂经动物代谢后产生含砷化合物及粪便经农用释放进入土壤[10]。历史上将砷类农药施用于水果作物后,在果园土壤中检测砷含量高达732 mg/kg。长期使用磷肥也会使土壤中富集砷。受工业污染的沉积物和土壤,其砷含量比自然条件下沉积物和土壤的本底值高几个数量级,达数千甚至上万mg/kg (表2)。
1.2 自然来源
含砷硫化物岩石和氧化物岩石经风化和雨水冲蚀等过程释放可溶态砷是土壤砷的主要自然来源。土壤和沉积物是最重要的砷汇,地壳中平均丰度约为1.5 mg/kg,土壤中砷含量约为5 ~ 15 mg/kg,全球土壤平均为7.2 mg/kg[8]。根据砷含量范围可将土壤沉积物和岩石分为3 类,第一类在5 mg/kg 以下,包括火成岩、变质岩、沉积岩中的砂岩以及河床沉积物等;第二类在5 ~ 15 mg/kg 范围,包括典型泥质岩(板岩、千枚岩)、沉积岩、泥质沉积物、河流沉积物、海洋黏土、煤矿、泥炭和黏土等,较高的含量反映了这类物质中存在一定比例的硫化物矿物、氧化物和有机物;第三类砷含量最高(20 mg/kg 以上),一般为硫铁氧化物,包括页岩、磷灰岩(达400 mg/kg)、铁质地层和富铁沉积物等(表3)。
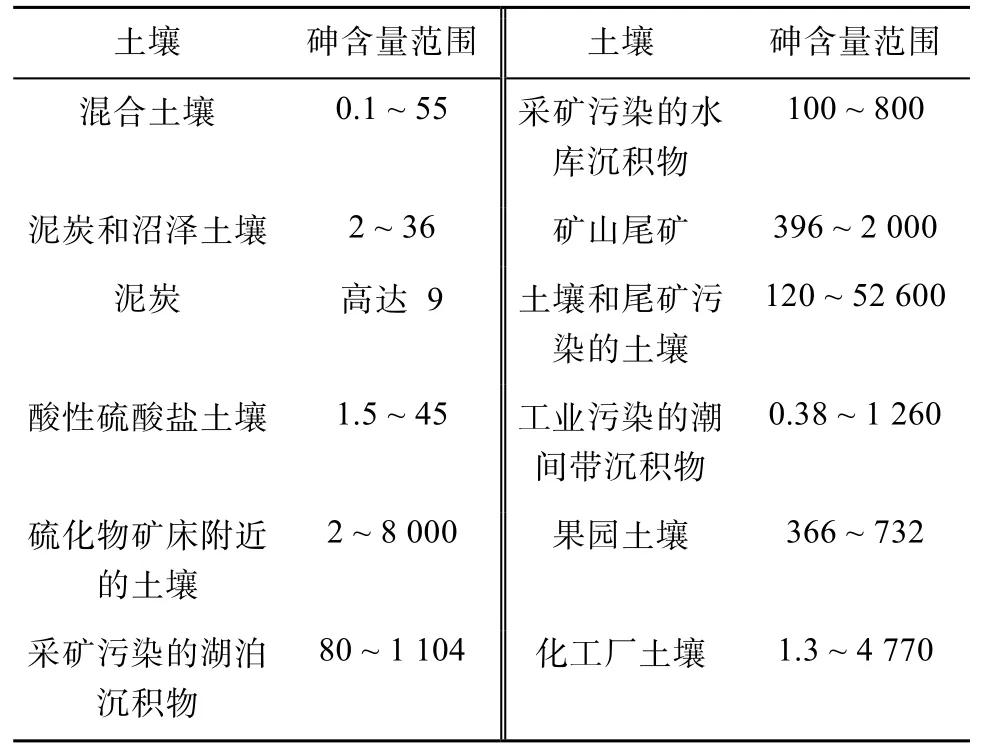
表2 土壤中砷的典型含量范围[8] (mg/kg)Table 2 Typical ranges of As concentrations in soils
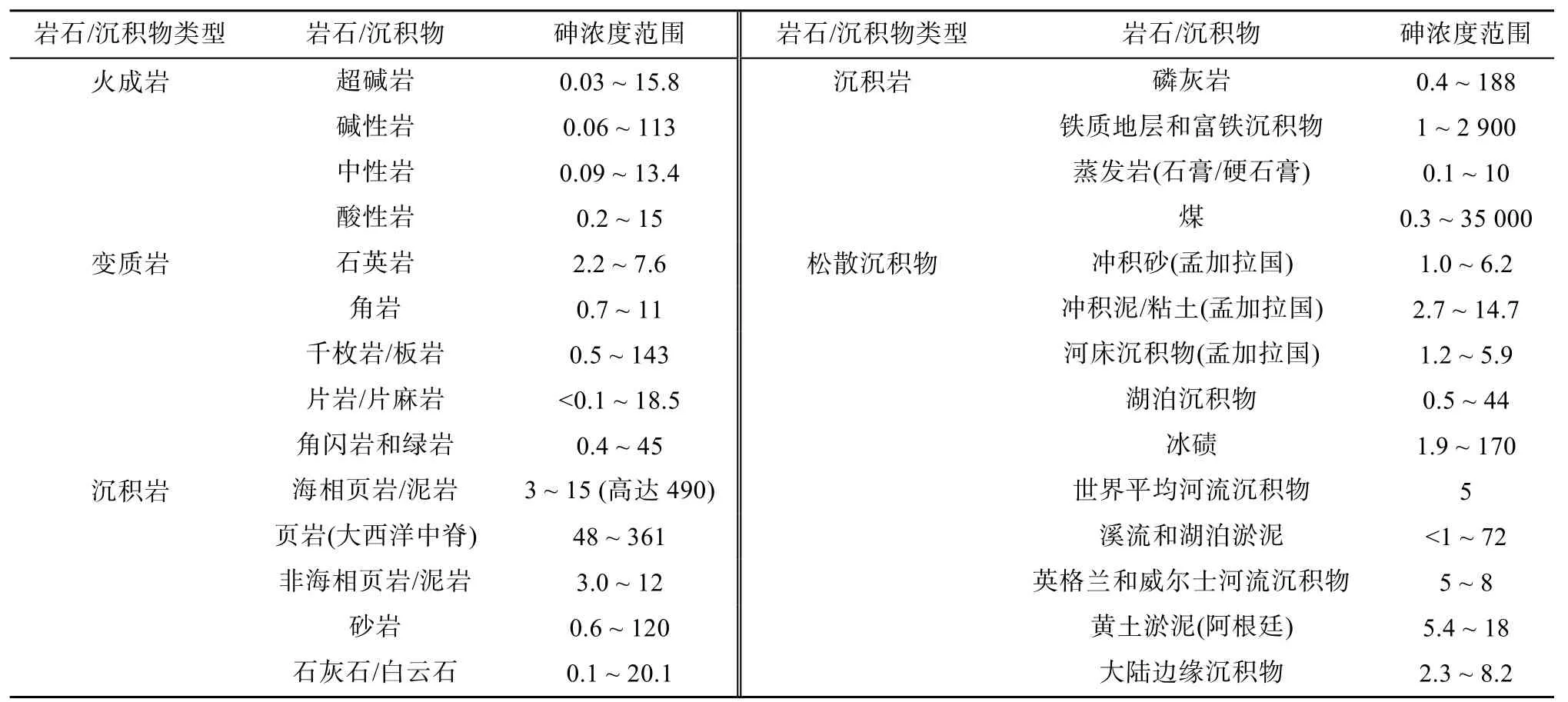
表3 岩石、沉积物中砷的典型含量范围[8, 11] (mg/kg)Table 3 Typical ranges of As concentrations in rocks, sediments
综上,土壤中砷的来源主要是人类工农业活动和自然岩石风化,自然来源相对贡献较小,相比之下,工业污染导致土壤中砷含量高出几个数量级。需指出的是,土壤中砷的含量水平、分布特征、砷的地球化学特性也在很大程度上与其所处的环境条件密切相关。
2 影响砷迁移的因素
2.1 金属矿物对砷迁移的影响
2.1.1 铁矿物对砷的影响 在自然体系中,铁矿物的类型和分布、还原条件的持续时间[12]以及铁矿物还原转化的速率和程度是决定砷固定或释放的主要因素。Fe(III)(羟基)氧化物还原释放砷被确定为导致土壤和沉积物中孔隙水砷浓度升高的罪魁祸首,已有大量文献报道了其相关性,如Huang 等[13]发现总铁与总砷具有显着的相关性(r=0.70,P<0.05);Bennett等[14]研究表明土壤溶液中Fe(II) 浓度和As(III) 浓度呈极显著正相关(r=0.896,P<0.001);进一步研究指出Fe(II)和总砷之间有显着的正相关性(r=0. 868,P<0.001),二者存在耦合释放的关系[15];Honma 等[16]研究也证实土壤溶液中Fe(II) 与总砷浓度存在二次函数关系:[As]= -0.002 4[Fe(II)]2+ 0.312 5[Fe(II)] +3.588 6。由此可见,铁矿物的还原性溶解和砷的迁移转化具有紧密的联系。
铁砷循环耦合关系及其机理主要包括两个方面,即铁砷耦合的氧化还原过程和铁矿物对砷的吸附解吸过程。1)铁砷耦合的氧化还原过程(图2)。铁砷耦合的氧化还原过程包括由铁还原菌的作用引起铁氧化物的还原溶解导致砷的释放和吸附在铁氧化物上砷的直接还原[17]。相比于土壤溶液中砷的还原,吸附在铁矿物表面上砷的直接还原半衰期极高,因此大量研究集中在铁矿物还原驱动砷释放。微生物介导的铁还原过程包括同化铁还原和异化铁还原过程,同化铁还原指Fe(III) 被运输到细胞体内被铁还原酶所还原;异化铁还原是指微生物将电子转移至细胞外的铁氧化物表面,将Fe(III) 还原成Fe(II) 的过程[18],其中异化铁还原是主要过程。在厌氧条件下,异化铁还原耦合砷氧化,Fe(III) 还原成Fe(II) 溶解,其反应式为CH3COO–+2Fe2O3+4Fe(OH)3+ 15H+→8Fe2++2HCO–3+14H2O;同时吸附的As(V) 进行溶解,溶液中As(V)浓度先增加,后在异化砷的作用下还原成As(III),其反应式为CH3COO–+2HAsO2–4+2H2AsO–4+5H+→4H3AsO3+2HCO–3[19]。异化砷是指氧化乙酸、有机质等电子供体耦合As(V) 还原,同时为微生物提供能量[20]。由于Fe(III) 和As(V) 同为电子受体,能替代铁还原菌或砷还原菌,因此二者之间存在竞争还原关系,在足够电子供体情况下铁砷先后被还原,但在电子供体不足的情况下,铁砷之间的还原就存在竞争(图3)。有机质能提高土壤溶液中砷的有效性[21],还能介导氧化还原电位的变化以顺利促使砷的活化[14]。而有机质耦合铁或砷的还原中,二者哪个更有耦合有机质降解的优势有待进一步研究。
2)铁矿物对砷的吸附解吸过程。在铁还原过程中发生3 个竞争过程,共同决定了As(III) 的溶解浓度:①孔隙水置换(流动)过程中化学失衡促进As(III) 解吸;②铁相的溶解释放As(III);③次生铁相固定As(III)[12]。故在砷解吸过程中也通过吸附、(共)沉淀或两个反应同时进行使砷重新固定[22]。先前存在大量报道铁(氢)氧化物还原溶解导致砷解吸释放的研究[14-16],但最近研究发现与之相反的情况,即随着水铁矿的还原转化,其重结晶为更稳定的次生Fe(II) 矿物相,增加了As(III) 和As(V) 的吸附固定[23],且进一步的研究表明水铁矿还原转化初始阶段增强As(III)固定(相对于非生物系统),而长时间的还原则导致砷的解吸[12]。这说明铁氧化物的还原溶解不一定会导致砷的迁移,Fan 等[24]也发现当As(V) 引入缺氧土壤时,能与Fe(II)/ Fe(III)快速共沉淀(形成砷酸铁和砷酸亚铁)阻止其还原为As(III),该过程是控制砷固定的主要机制。然而当砷对次生Fe(II)/ Fe(III) 矿物的结合能力较低时,吸附砷的Fe(III) 矿物的微生物还原则会导致砷的迁移。例如,在含As(III) 的Fe(III)矿物的微生物还原中,发现As(III) 强烈的迁移;但在含As(V) 的Fe(III) 矿物的微生物还原中则导致砷的固定,这种差异可归因于As(III) 和As(V) 与次生Fe(II)/ Fe(III) 矿物的结合能力的差异[25]。此外,铁还原菌FeRB 的活动可促进上述过程,一方面使含砷铁矿物和水稻根表铁膜[26]还原溶解而促进砷的释放,另一方面在铁还原过程中诱导形成对砷具有高结合能力的结晶次生铁矿物而促进砷的固定[25]。这些研究说明了铁氧化物的还原溶解对砷释放或固定的净效应取决于砷对次生铁矿物的结合能力。

图2 微生物铁还原耦合砷氧化过程Fig. 2 Microbial Fe reduction coupled As oxidation process
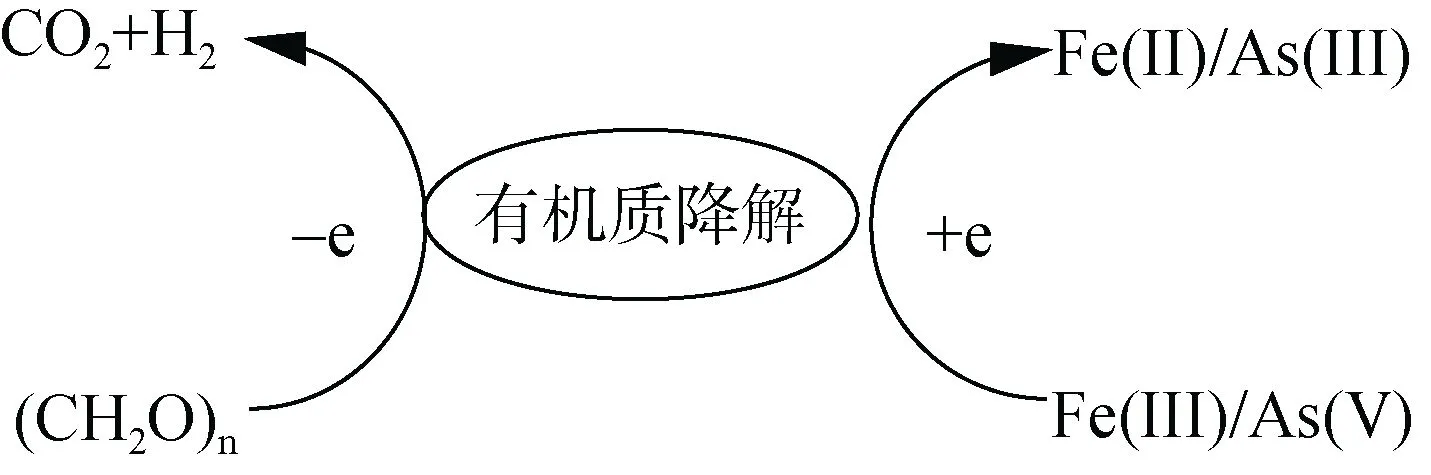
图3 铁砷竞争耦合还原Fig. 3 Fe-As competitive coupling reduction
沉积物和土壤环境中的铁矿物存在广泛的吸附砷过程[27],铁矿物吸持砷是一种有效的自然衰减过程,可显着降低高砷水体(如酸性矿山废水)中的砷浓度。例如,大多数土壤和沉积物中的铁矿物能与As(V)强烈结合,尤其是将其吸附到具有高比表面积的铁矿物上[28],如纤铁矿(β-FeO(OH)/γ-FeO(OH))和水铁矿(Fe5HO8·4H2O)(条件分别为2.5≤pH≤6 和6≤pH≤8)[29];而As(III) 的吸附更具特异性,其与Fe(III)(氢)氧化物结合最明显[30],并且在近中性和碱性的静水(间歇)条件下比As(V) 更广泛地吸附在铁(氢)氧化物和磁铁矿上[23,31]。不同类型的铁矿物对砷的吸附机制以及特点见表4。
2.1.2 铝氧化物对砷的影响 铝氧化物对砷的固定起着重要作用,其作用受到氢氧化铁强烈的干扰,因此研究通常将铝和铁综合一起讨论。Goldberg 和Johnston[41]研究了As(III)、As(V) 与铁、铝(氢)氧化物的相互作用并指出吸附机理是:As(V) 在铝(氢)氧化物或铁(氢)氧化物表面形成亲和力较强的内层吸附;而As(III)在铁(氢)氧化物表面同时形成内层和外层吸附,但在铝(氢)氧化物表面只能形成外层吸附,因此导致了As(III)极易从铝(氢)氧化物表面解吸。铝和铁对砷的吸附和释放影响机制如图4 所示。

表4 土壤铁矿物类型对于砷的吸附机制以及特点Table 4 Adsorption mechanisms and characteristics of As on soil Fe mineral types
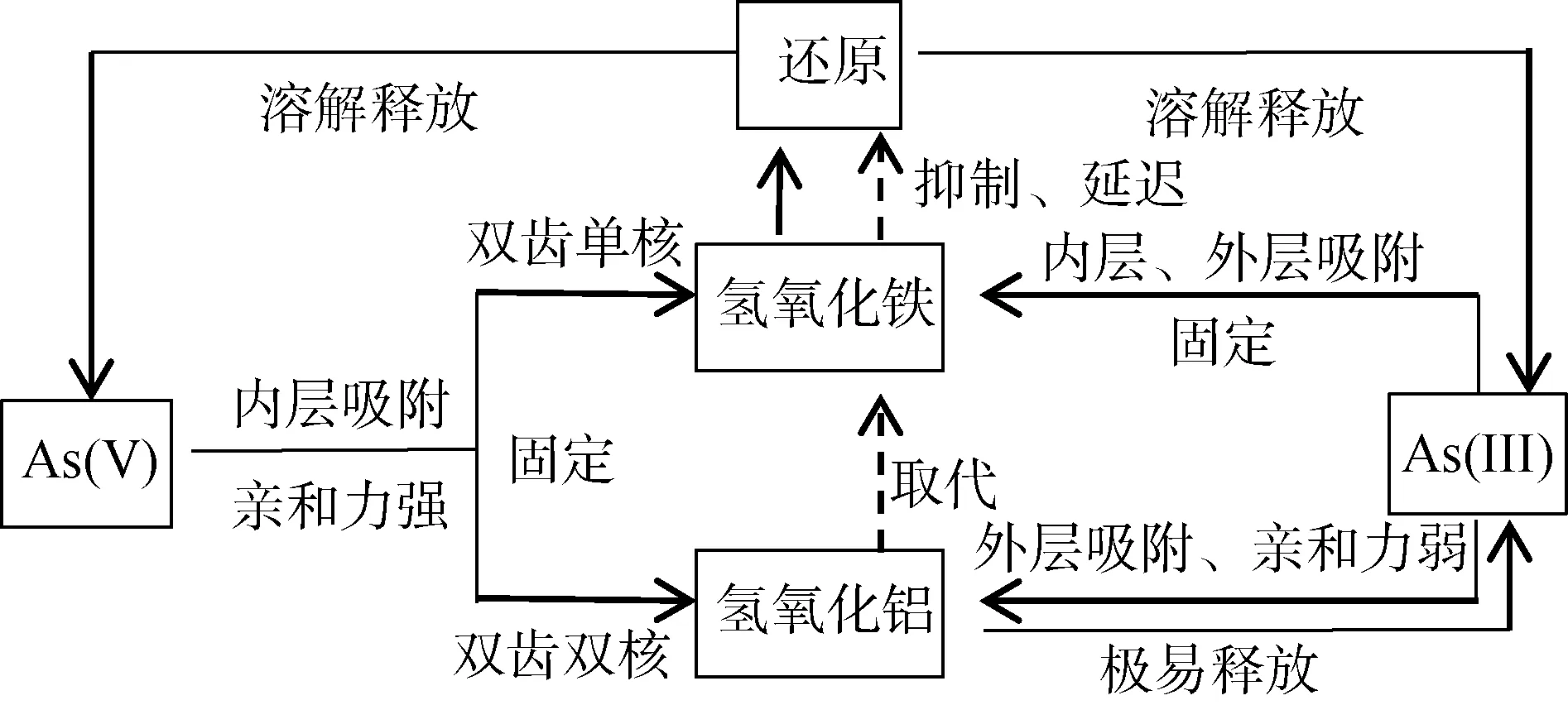
图4 铝铁氢氧化物对砷的吸附及影响机制Fig. 4 Mechanism of adsorption and influence of Al and Fe hydroxide on As
在pH<7.5、同等摩尔比的条件下,铝(氢)氧化物和铁(氢)氧化物对As(V) 的吸附能力大致相同;铝(氢)氧化物极易吸附As(V),吸附最大值在pH 4 ~4.5,而几乎不吸附As(III)[42],因此在任何pH 条件下,铁(氢)氧化物对As(III) 的吸附能力更强[43]。研究表明使用0∶1 或1∶4 的铝氢氧化物∶铁氢氧化物可以实现同等效果的最大As(V) 吸附率,而当铝铁摩尔比大于1∶4 时,随着其摩尔比的增加,形成结晶铝(氢)氧化物(三羟铝石和三水铝石)导致As(V) 和As(III) 的吸附量减少,尤其As(III) 的解吸增加[44]。因铝(氢)氧化物对As(III) 的吸附几乎可以忽略不计,故0∶1 的铝∶铁氢氧化物比1∶4 的铝∶铁氢氧化物吸附As(III) 效果更好[44]。进一步研究指出,铁(氢)氧化物介质中As(V) 还原形成的As(III) 可再次被铁(氢)氧化物吸附,向水相中的释放量非常低,但当铝引入后,As(III) 向环境的释放明显增加,尤其在铝(氢)氧化物体系中,几乎全部的As(III) 均被释放[45]。因此,铁(氢)氧化物比铝(氢)氧化物及两者混合物吸附砷的能力更强。
另一方面,As(V) 对于铝(氢)氧化物的键合力比铁(氢)氧化物更强。砷键合在结晶铝(氢)氧化物上是双齿双核,而砷对无定形羟基铁(氢)氧化物的键合是双齿单核[46];研究发现含铝/砷的土壤与砷铝石(AlAsVO4·2H2O)的EXAFS 光谱非常相似,其中砷以As(V) 形式存在,第一个As—O 配位壳体在1.70Å ±0.01Å 处填充4 个氧邻位,砷在3.18Å ± 0.02Å 处有2个铝邻位,这与双齿双核的As—O—Al 键(AsO4单元的两个O 原子分别键合到两个Al 原子上)一致[46]。此外,由于铁存在氧化还原过程[47],铁(氢)氧化物及其吸附的砷易溶解释放。土壤中大部分铁(氢)氧化物都被铝取代[44],共沉淀的铝/铁氢氧化物可能对于砷的吸持更有利,因为结构中存在铝取代的铁(氢)氧化物的还原被延迟,降低溶解速率[45]。如一项比较铝取代的针铁矿和纯针铁矿的细菌还原溶解研究证实,当在针铁矿中存在铝取代时,溶解速率更慢[48]。由此可见,在还原环境中,铝/铁氢氧化物混合物对于吸附砷可能比铁氢氧化物更有利。
2.1.3 锰氧化物对砷的影响 锰氧化物是一种非生物强氧化剂,已被证明可以氧化As(III)、Fe(II)、Co(II)、U(IV)、Cr(III) 和有机物质[49]。富锰的地层通常形成于地下水位频繁波动的区域,例如浅海区和浅层含水层[50]。冲积平原和泛滥平原中富锰物质的沉积过程也会产生富锰环境[51]。自然中重要的锰化物包括水钠锰矿物([Na,Ca,Mn(II)]Mn7O14·2.8H2O)、锂辉石(LiAl2[Mn(IV)2Mn(III)]O6(OH)6)(双层结构)、钡镁锰矿((Ca,Na,K)0.3~0.5[Mn(IV),Mn(III),Mg]6O12·3~4.5H2O)(隧道结构)[52],其零电荷低点(pH 1.7 ~ 3.5)使其成为各种重金属(Zn、Ni、Co 和Pb)的有效吸附剂[53]。锰氧化物的还原在热力学上比Fe(III) 和As(V) 更有优势,因其具有相对较高的氧化还原电位(约400 mV)[54]。锰化物具有与铁相似的氧化还原化学性质,在氧化条件下能迅速氧化 As(III) 和Fe(II),在土壤和沉积物的含氧/缺氧循环过程中与砷的固定/迁移有很强的联系[55]。锰氧化物对砷的影响过程如图5 所示。
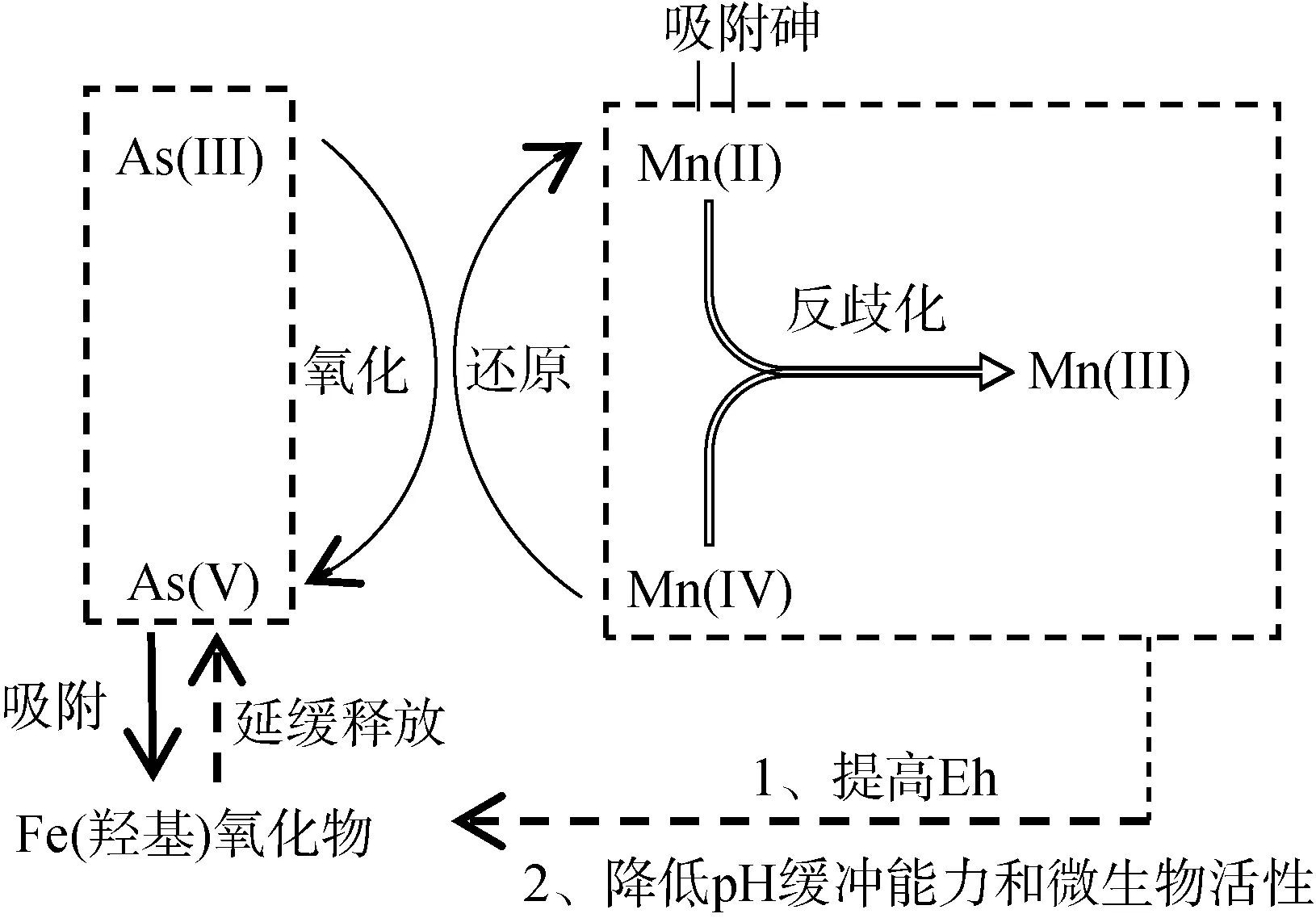
图5 锰氧化物对砷的影响过程Fig. 5 Effect of Mn oxide on As
锰氧化物将As(III) 氧化成As(V) 涉及多个同时反应,包括Mn(IV) 还原为Mn(II)、Mn(II) 吸附以及Mn(IV) 和Mn(II) 反歧化为Mn(III) 反应[56]。除了直接氧化As(III) 的作用外,锰氧化物还可以通过将氧化还原电位提高到更高的水平来延缓含砷的铁(羟基)氧化物的还原溶解以及砷向孔隙水的释放[57]。例如,在一个砷污染洪泛区土壤的研究中,Ehlert等[58]发现,水钠锰矿添加剂可促进Fe(II) 氧化形成新沉淀的Fe(III)氢氧化物,其作为As(III) 的高效吸附剂进一步延迟了砷和铁的还原,从而显著降低砷浸出率;Xu 等[59]也观察到了相同的结果,锰氧化物含量较高的土壤表现出较少的砷迁移特性,原因是锰氧化物能够减缓土壤淹水时氧化还原电位的下降,并使固相和溶液相中保持较高的砷酸盐/亚砷酸盐比率从而降低砷的迁移。此外,高浓度锰氧化物还可降低土壤还原期间的pH 缓冲能力,使微生物活性下降[58]。由此可见,富集氧化锰的土壤层能够有效降低砷的迁移。
但是,在微生物还原条件下锰氧化物对As(III)的氧化是有限的[56-57],微生物作用将As(V) 还原成As(III),由于锰的表面钝化反应,As(III) 不再被水钠锰矿或Mn(III) 的转化产物所氧化。尽管也有研究发现亚锰酸盐也可氧化As(III)[60],但是贡献不大。表面钝化反应可能是由于水钠锰矿转化为亚锰酸盐的缓慢动力学导致Mn(II) 和Fe(II) 吸附在水钠锰矿表面,分别导致还原归中反应(Mn(IV) 和Mn(II) 反歧化为Mn(III))和表面沉淀[58];另一原因是微生物代谢产物积累覆盖了锰氧化物活性表面。因此锰氧化物对As(III) 的氧化能力在淹水土壤和沉积物中可能受到限制。在这种环境下,吸持在固相中的As(III) 的稳定性是短暂的,进一步的土壤还原将导致次生Fe(III) 相的异化还原,最终导致As(III) 和Fe(II) 的释放。但是,在氧化还原条件频繁变化的情况下,富含锰氧化物的土壤可能成为长期的砷汇,使砷浸出风险降低,促进富锰氧化物的土壤层中砷的富集。因此,锰氧化物是调控污染土壤中砷浸出风险的重要因素之一,在治理砷污染方面很有前景。
2.2 阴离子对砷迁移的影响
2.2.1 硫循环对砷的影响 砷可形成不溶性硫化物矿物,包括雌黄(As2S3)、雄黄(AsS)和毒砂(FeAsS),也可形成多种金属硫化物,包括四方硫铁矿(FeS)、胶黄铁矿(Fe3S4)和黄铁矿(FeS2),并可在这些硫化物上作为吸附络合物存在[61]。硫化物矿物的形成通常通过提供电子供体以及在某些情况下向沉积物和土壤中添加无机硫酸盐来刺激土壤中硫酸盐还原菌(SRB)的活性来实现。通过该过程,溶解态的砷可以形成含砷硫化物固体[61]。微生物硫酸盐还原一般不直接影响溶解态砷浓度[62],而硫酸盐化学还原形成的硫化物可以通过共沉淀与吸附共同作用降低砷浓度[63]。另一方面,地下水中常见的硫氧化剂包括溶解氧、硝酸盐和Fe(III)(低pH 值时)可以使含砷硫化物再次氧化[64],促进砷释放到地下水中。根据已发表的关于硫酸盐还原的研究,提出一个描述硫循环过程的概念模型,并分析该过程固定和释放砷的机理(图6)。
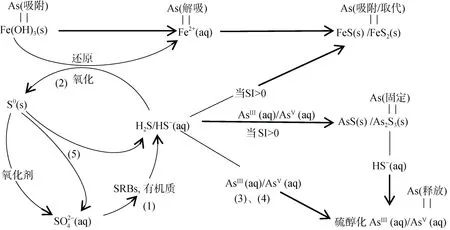
图6 环境中铁和硫氧化还原循环以及不同相之间的砷分配[65-69]Fig. 6 Fe and S redox cycle in the environment, and As partitioning among different phase
在存在乳酸盐的硫酸盐微环境中,初期存在硫酸盐还原菌(SRBs)的微生物滞后期,然后硫酸盐还原可以与Fe(III) 还原相当的速度进行,甚至由于动力学原因在某些阶段取代Fe(III) 还原过程[65]。硫酸盐被SRB 还原成硫化氢 (HS–和H2S 的混合物,pKa,H2S=7.02):SO+ 2C3H5O↔HS-+ 2HCO+ 2C2H3O+H+(式1)。当铁和砷硫化矿物达到饱和(即饱和指数> 0)时,游离硫化物(HS–)反应形成不溶性FeS 和砷硫化矿物(图6),FeS 的形成可使土壤孔隙水中Fe(II) 浓度降低,而四方硫铁矿的形成耦合As(V) 还原为As(III),As(III)进一步被As2S3类物质螯合[61]。如果存在活性Fe(III) 矿物质(如水铁矿),则游离硫化物与其发生还原反应并产生Fe(II) 和零价硫(可能还有少量的硫代硫酸盐):HS-+2Fe(OH)3(S)+ 5H+↔2Fe2++S0(s)+ 6H2O(式2)。这个涉及电子转移过程的反应由表面控制,是一个快速的化学过程,水铁矿还原形成Fe(II),进一步转化为针铁矿和磁铁矿[66],因表面积减少而导致砷从土壤沉积物中解吸。此外,砷的还原过程是砷与硫化物形成共沉淀的必要步骤[67],游离硫化物不仅可以还原活性Fe(III) 矿物质(式2),而且可以还原砷酸盐[68]:H2AsO+ 3HS–+ 2H+↔HAsS3O2–+3H2O(式3),HAsS3O2–+ HS–↔AsS+ H2O(式4),通过形成溶解的硫醇化砷化合物如HAsS3O2–和AsSO3–4而增加砷的溶解度[69]。
零价硫的存在是硫酸盐还原的诊断指标,因为它只能通过硫化物氧化形成(式1、式2)。然后零价硫可能会由微生物介导发生歧化反应,再次产生硫化物和硫酸盐[67]:4S0(S)+4H2O↔3HS–+ SO+ 5H+(式5)。
这一系列过程(式1、式2 和式5)形成了有效的生物地球化学硫循环,催化了Fe(III) 还原,并以相对低的水平缓冲溶解的硫化物浓度,直到活性Fe(III)被耗尽。该过程说明硫酸盐还原产生的硫化物能够还原Fe(III) 和As(V),促进砷释放到水相[70],但又能通过促进砷和铁硫化物矿物的沉淀而固定砷[71]。此外,不同介质产生的铁水合物对砷的吸附能力显著不同:由硫酸盐培养基制备的铁水合物具有高于硝酸盐所制备的砷吸收容量[72],这说明了硫酸根离子能参与Fe(III) 水解,并且改变了所形成的水铁矿的表面化学性质,使其更有利于砷酸盐的吸附。另一方面,与吸附过程相比,As(V) 与Fe(III) 的共沉淀可产生更高的砷去除率,可能是因为在特异性吸附阴离子(即AsO)存在下吸附剂的活性位点和表面积最大化,并在共沉淀过程中形成了无定形的砷酸铁使可溶性砷的量最小化,因此总体上有助于显著降低砷溶解度。由此可见,微生物硫酸盐的还原可以影响砷在土壤中的归宿[61]。
如果硫循环过程中不存在活性Fe(III) 矿物质这种易氧化硫的氧化剂,则保持低活性的硫循环过程会被破坏,因此在相对还原的体系内(如填埋场),更有利于不溶性硫化物的形成和砷的固定。因此,关于密闭基岩含水层中硫化物氧化导致砷污染的报道并不多见,因为完全饱和的含水层中存在的硫化物由于还原环境和缺乏氧化剂而基本保持稳定。因此,厌氧条件下硫化物对砷的释放迁移贡献较小,但高砷含量的黄铁矿和白铁矿发生氧化则易导致砷的大量释放。
2.2.2 磷酸盐对砷的影响 尽管磷酸盐(Pi)化合物经常被用于促进金属如铅和锌的固化,降低其在土壤中的迁移率和生物利用度[73],然而在砷污染土壤中添加磷肥却能增加砷的溶解度和流动性,促进植物对砷的吸收[74]。土壤中的砷主要以无机形式(砷酸盐As(V)和亚砷酸盐As(III))存在,有机形式的二甲基砷酸(DMAA)和一甲基砷酸(MMAA)的含量相对较小[75]。在有氧条件下,无机砷和Pi 由于相似的化学特性能够形成稳定的+5 价氧化态四面体氧阴离子[76]。由于两者物理和化学的相似性,As(V)和Pi 离子在相同土壤带电表面存在强烈的相互竞争。在相同的Pi 和砷浓度下,由于Pi 阴离子具有更小的尺寸和更高的电荷,且不易与溶解的硫化物反应形成不溶的矿物质或溶液配合物,因此比As(V) 结合到土壤上更具优势[77],从而更多的砷因吸附受到抑制而留存在溶液中被植物吸收。例如,Okkenhaug 等[78]发现,使用含磷的营养物质可以促进土壤溶液中砷的起始浓度由1 400 μg/L增大到1 700 μg/L;MorenoJiménez 等[79]也报道了,在Pi 存在的情况下增加了砷在土壤中的迁移。从另一角度,Tu 和Ma[80]发现添加磷可提高砷的生物利用度,施用磷对于使用蜈蚣草植物修复砷污染的土壤具有显著的增强效果。此外,Pi 对土壤的吸附缓慢,随着时间的推移将增加竞争As(V) 的吸附作用[76]。根据Steindorf-Rebhun-Sheintuch 方程、配体交换理论和共享电荷假设,Pi 有很大的可能取代土壤吸附的As(V)[81]。在使用飞灰改良的土壤中,Qafoku 等[82]报道了Pi 与砷酸盐竞争全部可用的非特异性和特异性吸附位点,取代了As(III) 和As(V),增加了土壤中砷的流动性。
砷和Pi 的竞争吸附作用与土壤成分的性质有关,如砷酸盐吸附量随着土壤阴离子吸附量的增加而增加[83]。Violante 和Pigna[84]发现当As(V) 和Pi 以等摩尔比供应时,富铁、锰矿物(如针铁矿、绿脱石、含铁绿土、水钠锰矿和软锰矿)吸收的As(V) 比Pi更多;然而无定形铝矿物上(如三水铝石、勃姆石、水铝英石和黏土部分包括高岭石、伊利石和蛭石等)吸附Pi 比As(V) 更多;对于任何pH 的石灰性紫色土壤,As(V) 和Pi 的吸附和解吸特性相似,遵循Langmuir 和Freundlich 方程[85]。在Bolan 等[83]的研究中,磷对砷的吸附和解吸影响取决于土壤阴离子的吸持能力,弱阴离子吸持能力的非水铝英石土壤对砷的吸附量远小于强阴离子吸持能力的水铝英石土壤的砷吸附量,而磷的添加明显促进了水铝英石土壤中砷解吸,非水铝英石土壤中添加磷对土壤砷的解吸和生物有效性的影响几乎可以忽略。Peryea[86]也指出砷的解吸依赖于土壤类型,只有大量添加磷(P > 400 mg/kg)才会影响火山土壤中砷的溶解度。当土壤中Pi 含量非常高时,含阴离子土壤和碱性土壤中砷的迁移率和溶解度都会受到强烈的影响。
2.2.3 硅酸盐和碳酸盐对砷的影响 硅和碳是同族元素,硅酸根(SiO)和碳酸根(CO)对砷的竞争吸附机理相似。SiO具有与AsO3–3相似的空间结构,因此能够同AsO竞争铁矿物表面的吸附位点,促进SiO和HSiO从铁矿物释放。例如,Robins 等[87]指出,硅酸盐能吸附在固体水铁矿和铝铁矿的表面上,与Fe(III)发生络合,从而导致As(V) 解吸;Swedlund和Webster[88]的研究也表明,硅酸盐可抑制As(III) 和As(V) 在水铁矿上的吸附。目前许多研究都忽略了碳酸盐对吸附态砷酸盐和亚砷酸盐的取代作用,实际上地下水中碳酸盐含量通常较高,且碳酸盐吸附较强,可改变零电荷点(PZC)、Zeta 电位和氧化物的质子缓冲能力[89],能够极大地影响砷的吸附。在自然环境下,吸附大量砷的沉积物存在于低浓度碳酸盐的地表水中,当这些沉积物暴露于高含量的溶解碳酸盐的地下水时,沉积物表面的砷就会因被CO取代而发生迁移。Appelo 等[90]使用表面络合模型计算,证实了土壤和地下水中沉积物的吸附特别是碳酸盐的吸附能够显著降低砷在水铁矿上的吸附能力。这些研究说明硅酸盐和碳酸盐都是环境中砷浓度升高的促进因素。
2.3 土壤理化性质对砷迁移的影响
2.3.1 土壤氧化还原电位(Eh)对砷的影响 Eh 的周期性变化是水稻土独特的特征,其变化范围通常在+200 ~ +700 mV[91],这种广泛的变化直接导致了水稻土中一些氧化还原敏感物质如氧、铁、锰、氮、硫和碳的剧烈变化。淹水前,土壤孔隙结构可能充满空气,土壤环境的Eh 在300 mV 以上;淹水后,土壤孔隙网络中的游离氧被微生物快速消耗,Eh 迅速降至200 mV 以下,此时微生物依次使用NO,Mn4+,Fe3+,SO作为电子受体进行还原,伴随着微量气体NH3,N2O,N2,H2S,CH4的排放以及pH 的上升[92],形成强烈还原的缺氧环境,土壤固相铁矿物和As(V)均被还原溶解;大部分水稻收获前一到两周进行田间排水,Eh 又上升到400 mV 以上,还原态化合物如Fe(II) 和As(III) 被氧化Fe(III) 和As(V),As(V) 与铁氧化物结合,含量及有效性均明显降低[93]。
长期水淹条件会导致土壤Eh 降低,释放到溶液中的 As(III) 和总砷的浓度均显著提高。例如,Yamaguchi 等[94]指出当土壤Eh 从 +500 mV 降至+100 mV 时,土壤溶液中As(III) 浓度和Fe(II) 浓度显著增高;Yamaguchi 等[95]发现,水淹条件下当水稻土的Eh 由+100 mV 下降至 –68 mV 与 –75 mV 时,溶液中砷的释放量每千克土分别提高了6.9 μmol 和19 μmol;Syu 等[96]报道了砷污染的水稻土经历淹水后土壤溶液砷含量大幅度上升至3 000 μg/L 以上,伴随着Eh 的急剧下降与Fe(II) 含量的快速上升;进一步研究指出砷浓度与Eh 存在关系可表示为[As]=5.84exp(–0.0145 Eh)[16]。这些研究说明土壤砷的释放量和土壤Eh 值呈显著负相关关系。然而,当淹水70 d 后,水稻土壤溶液中的砷浓度趋于保持稳定[97],可能是由于长期淹水使水稻土的pH 保持相对稳定的动态平衡和氧化还原态平衡,从而砷也保持在土壤和水之间的分配平衡。此外,反复氧化还原循环可以降低还原过程中高达45% 的砷迁移率[98],其原因可能是Eh升高时形成的次生铁矿物α-FeOOH/Fe(OH)3与砷共沉淀。由此可见,土壤经历淹水缺氧过程导致Eh 下降,砷的迁移率增加,但长期淹水或反复淹水可能保持一定的As(III) 含量不变甚至降低其迁移率。
2.3.2 土壤pH 对砷的影响 土壤pH 是影响砷形态和有效性的关键理化性质之一。厌氧条件下由于微生物对有机质的降解可导致pH 降低,这种酸性条件有利于矿物表面的质子化、砷酸盐阴离子(H2AsO–4或HAsO2–4)的吸附以及降低As(V) 的溶解度,提高亚砷酸(H3AsO3)的溶解度。在碱性条件下,As(III) 优先于As(V) 吸附在氧化铁表面上;在近中性的pH,即接近H3AsO3的pKa(9.2),砷酸盐和亚砷酸盐都通过形成强内层络合物在水合氧化铁(HFO)和结晶氧化铁的表面处以相同的效率吸附[99]。Honma 等[16]研究表明,溶解性砷浓度与pH 存在关系可表示为[As]=3.56×10–12exp(4.72 pH);相似的研究也指出总砷和pH的关系式为[As]= –0.557+3.61×10–11exp(3.50 pH)[15],表明土壤砷的释放量和土壤pH 呈正相关。这些研究说明了水稻土微环境中Eh 降低和pH 升高是驱动砷释放的关键因素。
2.3.3 有机质对砷的影响 土壤有机质是铁代谢微生物和矿物质相互作用过程的重要参与者,它的氧化能够直接耦合土壤沉积物和溶液中砷的微生物还原[100]。有机化合物尤其是可提供离子参与形成Fe(III)络合物的有机物,会严重干扰砷酸盐在水铁矿和相关吸附剂上的吸附,如黄腐酸(FA)或腐殖酸(HA)吸附在针铁矿表面可导致砷酸盐的解吸,增加溶液中砷浓度[101]。一方面,有机质中的羧基(–COOH)和羟基(–OH)官能团能够与金属氧化物发生配位体交换[78],提高土壤溶液中砷的有效性[21];另一方面,有机质的微生物降解通常耦合Fe(III) 矿物还原过程,进而改变土壤固相或土壤溶液的平衡状况,促进土壤固相中砷的解吸[21]。Fe(III)和As(V) 同为电子受体,二者之间可能存在竞争还原与氧化关系,在足够电子供体情况下铁砷先后被还原,但在电子供体不足的情况下,氧化还原过程中铁和砷之间可能存在竞争,如图7 所示。
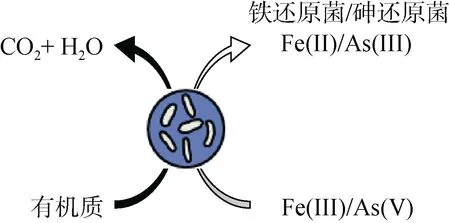
图7 有机质参与砷还原过程Fig. 7 Organic matter involved in As reduction process
有机阴离子促进As(V) 释放能力大小为柠檬酸盐>草酸盐>>乙酸盐[102],环境中竞争性物质对砷吸附的影响顺序为磷酸盐>腐殖酸>硅酸盐>硫酸盐[103],其干扰作用(尤其是腐殖酸)在较碱性pH 时增加。腐殖酸和磷酸盐、硅酸盐不仅通过竞争性吸附降低砷的吸附,还能与Fe(III) 氢氧化物形成复合物,而硫酸盐主要通过竞争性吸附抑制砷的吸附,因此前者抑制砷的吸附作用比硫酸盐更强。此外,通过提供更高的电子供体(如乳酸盐)浓度可提高Fe(III) (氢)氧化物还原速率,从而加速As(III) 的解吸[12]。由此可见,有机质也是影响砷还原释放的关键因素之一。
值得注意的是,砷的释放程度并不总是与固体表面吸附其他物质的量相匹配,因而砷的释放不能仅仅用竞争吸附/解吸来解释。砷的解吸应通过砷的竞争吸附/解吸、铁和铝的氧化/氢氧化物的溶解以及Fe(III)和Al(III) 与As(V)、OH–、柠檬酸盐、草酸盐、硅酸盐、磷酸盐、以及pH 和Eh 等多种因素的复合来解释砷解吸的复杂机制。
3 结论与展望
砷具有独特的物理和化学性质,在环境中的迁移过程是通过生物地球化学反应与人类相互作用的复杂结合而发生的,受微生物、铁、铝、锰、硫、磷、硅酸盐、碳酸盐、pH、Eh 和有机质等诸多因素的影响。对文献的分析表明,造成砷向环境迁移的主要原因是铁锰氧化物矿物的还原溶解、黄铁矿等硫化物矿物的氧化溶解、铝氢氧化物表面的砷还原、有机质抑制砷吸附以及Eh 降低和pH 升高。未来的对砷深入研究应集中在下列3 个方面:
1)探讨上述因素在不同性质土壤中的相互作用以及促进砷释放的相对贡献值。砷的释放可能通过上述多种因素的复合,作为pH 和Eh 的函数来表达砷解吸的复杂机制;应重点考虑铁锰氧化物以及碳酸盐,目前有关锰氧化物对污染土壤中As(V) 还原和释放影响的研究较少,碳酸盐是研究砷时最易忽略的一个因素,而地下水中碳酸盐含量通常较高且对砷的吸附影响作用较大。此外,砷在全球土壤时空分布规律以及导致当地高砷含量的因素、降低当地砷污染的对策有待进一步研究;目前已有许多研究报道了当地地下水砷的分布规律及迁移释放的影响因素,然而对于这方面内容在全球地下水中缺乏综合性分析。
2)进一步研究厌氧条件下铁砷还原与氧化过程的动力学机制以及铁砷耦合的主要影响因素。目前有关铁参与砷的形态变化和迁移转化研究较多,然而控制铁还原过程的关键因素仍不清楚。Fe(III) 与 As(V)同为电子受体,两者间的竞争性也缺乏研究。研究证实硝酸盐依赖性细菌 FeOBAcidovoraxsp. BoFeN1在添加 As(III) 的时候,系统中的铁矿物被还原,说明在微生物体内确实存在铁砷耦合的反应机制,然而微生物在铁砷耦合作用过程中的生物化学机理有待进一步研究。
3)土壤中砷的氧化还原转化与微生物息息相关,砷氧化微生物可分为化能自养型砷氧化微生物(CAOs)、异养型砷氧化微生物(HAOs)两类,CAOs 在厌氧环境中由硝酸根作为电子受体。HAOs 同样也能够氧化As(III),但是需要有机物质作为能量与细胞物质的来源。目前,砷代谢微生物研究主要集中于可培养菌群对砷的迁移转化的影响及其机理的研究,环境中不可培养的砷代谢微生物的生物多样性和群落结构有待进一步的研究。
