苋菜红表面增强拉曼光谱的密度泛函理论研究
2020-06-13逯美红雷海英王志军张竺立程旭丽吴艳波
逯美红,贾 娟,雷海英,王志军,张竺立,程旭丽,吴艳波
1.长治学院电子信息与物理系,山西 长治 046011 2.内蒙古工业大学理学院,内蒙古 呼和浩特 010000 3.长治学院生物科学与技术系,山西 长治 046011 4.山西大学分子科学研究所,教育部化学生物学与分子工程重点实验室,山西 太原 030006
引 言
苋菜红又称为鸡冠花红、萘酚红,属于偶氮类磺酸型水溶性人工合成色素,可以用于食品、药品、化妆品等的着色。而人工合成食品色素多由煤焦油、苯、萘等芳香化合物为原料制成,若过量添加会导致基因突变甚至致癌,威胁人类健康[1],所以对苋菜红的检测显得尤为重要。
目前,常用的检测方法有高效液相色谱法[2]、示波极谱法[3]、光谱扫描法[4],这些方法大都具有操作繁杂、实验条件苛刻、样品制备复杂、对样品损害性高等不利因素。因此,亟需一种操作简单、准确性很高的技术来检测人工合成色素。拉曼光谱技术通过指认分子的特征峰识别与鉴定分子结构,已在化学、农学和医疗方面得到广泛的应用[5-7],但对于低浓度痕量色素检测通常需要进行表面增强,这就是表面增强拉曼光谱技术。庄志萍等对除草剂西玛津表面增强拉曼光谱的密度泛函理论进行了研究[8],邹乔等对菲分子结构与光谱进行了密度泛函理论研究[9],陈玉锋等对杀菌剂三环唑的表面增强拉曼光谱进行了研究[10],但对人工合成色素苋菜红分子的表面增强拉曼光谱的实验和理论研究还未见报道。
从实验与理论两个方面对苋菜红分子的常规拉曼光谱与表面增强拉曼光谱进行研究,通过对苋菜红分子的前线轨道、静电势与极化率的计算和分析,探究该分子与Ag原子配位的合适位置。并在此基础上分别对苋菜红分子与1个Ag配位的复合物(Ag1-Amaranth)及与3个银原子团簇配位的复合物(Ag3-Amaranth)的表面增强拉曼光谱进行研究。
1 实验部分
1.1 方法
实验中所用样品购自生工生物(上海)股份有限公司,粉末状。分子式为C20H11N2Na3O10S3,分子量为604.3。实验测试所用仪器为显微共聚焦拉曼光谱仪(德国Bruker)。实验测试的条件为:激发波长为532 nm,激光功率为20 mW,分辨率为2~3 cm-1,在室温下对苋菜红粉末进行检测。
1.2 计算方法
采用GaussView 5.0和Gaussian09软件[11-12],在密度泛函理论B3LYP/6-31++G(d,p)基组水平上搭建分子结构优化并进行计算,计算了苋菜红分子的前线轨道、静电势及极化率,探究苋菜红分子与基底Ag原子的最佳配位位置。并且对苋菜红分子配位1个Ag原子及3个Ag原子团簇形成的复合物在B3LYP方法下使用6-31++G(d,p)(C,H,O,N,S,Na)基组以及Sdd(Ag)基组进行结构优化及表面增强拉曼光谱的计算。
2 结果与讨论
2.1 苋菜红分子结构优化
基于密度泛函理论,优化分子结构并进行振动频率计算,所得振动频率均大于0,结果无虚频,说明优化后的分子结构为稳定构型,优化后的最小能量为-3 312.06 eV,其优化后的分子结构如图1所示。

图1 优化后的Amaranth分子结构Fig.1 Optimized molecular structure of Amaranth
2.2 前线轨道计算
前线轨道即最高占据分子轨道(highest occupied molecular orbital,HOMO)和最低未占分子轨道(lowest unoccupied molecular orbital,LUMO)是量子化学的一个重要参数。HOMO轨道是能量最高的电子填充轨道,所受束缚最小,最易失去电子;LUMO轨道是能量最低的未填充电子的空轨道,易接受电子。因此前线轨道决定着分子的电子得失和转移能力,决定着分子间反应的取向等重要化学性质,其能量及能级差对研究分子的化学性质有重要意义[13]。计算结果表明,第153号分子轨道为HOMO轨道,E153=-5.972 95 eV,第154号分子轨道为LUMO轨道,E154=-3.057 23 eV,能隙ΔE=E154-E153=-2.915 72 eV,能隙很小。表明电子容易从HOMO跃迁到LUMO。为了全面进行分析电荷密度分布情况,选择对152轨道,153轨道,154轨道,155轨道分别计算电子云及分布见图2。由图2可以看出,苋菜红分子的HOMO,LUMO及邻近轨道上的电子云多数呈局域分布。HOMO与LUMO轨道电子云主要分布在偶氮基团与两侧的萘环结构上,在磺酸钠基团(—SO3Na)处都没有电子云分布,电荷密度低,说明—SO3Na基团对苋菜红分子的拉曼光谱特性影响较小,反应活性主要集中于苋菜红分子偶氮基团结构处。
2.3 静电势计算
分子静电势是判断分子反应活性和识别分子的有效途径。其不同颜色部位代表分子静电势定性大小,由此可以判断分子结构与基底或其他金属原子的配位或吸附情况。利用Gaussian09软件计算得到苋菜红分子的静电势如图3所示。红色区域表示为负电荷或亲电区域(电子的密集区域),容易受到亲电试剂的进攻;蓝色区域表示为正电荷或亲核区域,容易受到亲核试剂的进攻。从图中可以看出,若金属Ag原子与苋菜红分子配位,Ag原子应该选择蓝色区域处—SO3Na基团附近配位,但由于该基团变形性小,理论上形成稳定的包络面不宜配位,所以,苋菜红分子配位最佳位置是比—SO3Na基团附近电负性稍弱的区域。苋菜红分子的萘环结构与偶氮基团为一共轭体系,电子在整个体系上进行平均化分布,从而在偶氮基团处观察到静电势为浅蓝色,相对权衡之下,选择在电子密度较大,电负性强且电子流动性较大的偶氮基团处进行配位。
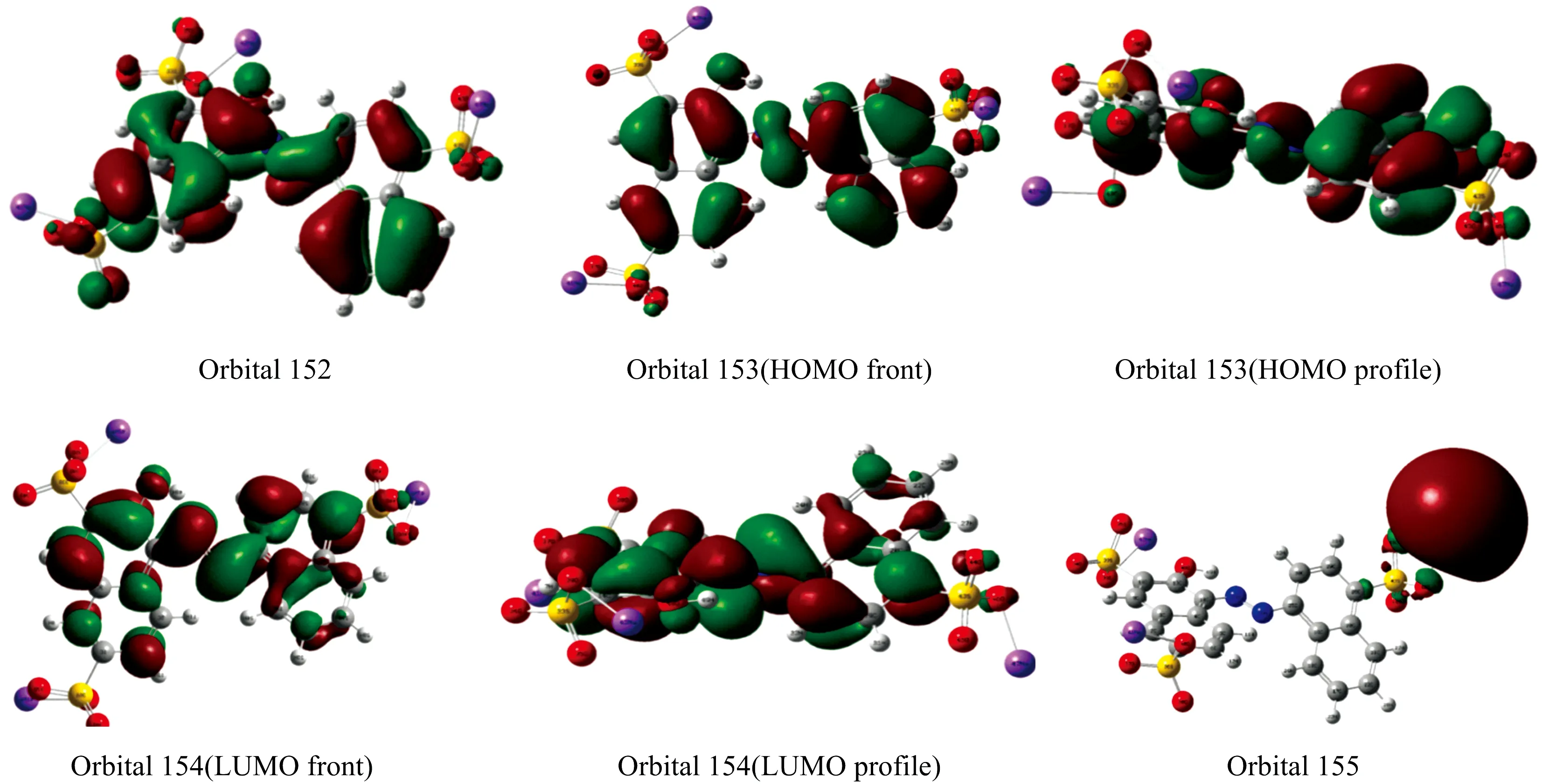
图2 苋菜红轨道电子云分布图Fig.2 Molecular orbital densities of Amaranth

图3 苋菜红分子表面静电势Fig.3 The surface electrostatic potential of amaranth molecular
2.4 极化率及自然键轨道布局分析计算
极化率及自然键轨道布局分析可以表征分子中的电子受约束及电荷转移情况,也是基于电荷转移模型表面增强效应中化学增强的的有力证明。基于以上前线轨道、静电势的计算与分析,选择苋菜红分子中距离偶氮基团2.33 Å附近处分别配位1个Ag原子及3个Ag原子团簇形成复合物。分别计算其极化率及自然键轨道局部分析,结果分别如表1和表2所示。从表1可以清楚的看出,分子极化率三个分量αxx,αyy,αzz逐渐增加,说明电子在金属Ag原子和苋菜红分子之间发生了转移,从而改变了分子极化率,发生了拉曼增强效应。从表2中也可以看到,随着苋菜红分子配位Ag原子个数的增多,配位处N原子的电荷数由-0.060增加到-0.459,与其直接配位的Ag原子电荷数由0.013 4增加到1.842,说明该N原子的电负性增加,更易与Ag发生电荷转移而导致化学增强。

表1 苋菜红、苋菜红-Ag1和苋菜红-Ag3复合物极化率Table 1 Polarizability of Amaranth,Amaranth-Ag1 and Amaranth-Ag3 compounds

表2 苋菜红、苋菜红-Ag1和苋菜红-Ag3复合物N和Ag的NBO电荷分析Table 2 NBO charge analysis of Amaranth,Amaranth-Ag1 and Amaranth-Ag3 compounds
2.5 苋菜红分子表面增强拉曼光谱的计算
在密度泛函理论下,对C,H,O,N,S,Na原子用基组6-31/G++(d,p)以及对Ag原子用基组Sdd进行拉曼光谱计算,优化后的分子结构如图4所示。

图4 优化分子结构图Fig.4 Optimal molecular structure diagram
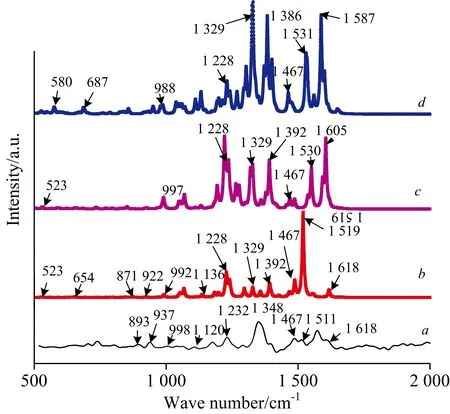
图5 表面增强拉曼光谱对比图Fig.5 Comparison of surface ehanced Ranman spectra(a):Experiment result;(b):Amaranth; (c):Amaranth-Ag1;(d):Amaranth-Ag3
图5a和b分别代表苋菜红分子的实验拉曼光谱图与理论拉曼光谱图。由图5a可以看出,在1 232,1 348,1 467和1 511 cm-1处有明显的拉曼特征峰,与理论拉曼的特征峰1 228,1 329,1 467和1 529 cm-1相对应,实验与理论结果整体吻合较好,可以作为鉴定和识别这种物质的拉曼特征指认峰。图5b,c和d分别为Amaranth,Amaranth-Ag1,Amaranth-Ag3的拉曼光谱图。可以看出,随着加入的配位Ag原子数目的增多,拉曼特征峰有两种变化情况,一方面拉曼特征峰个数有增多;另一方面拉曼特征峰对应的强度有加强。且随着偶氮基团处配位Ag原子的个数增多,对应拉曼光谱的峰位强度逐渐增强。振动模式归属见表3。
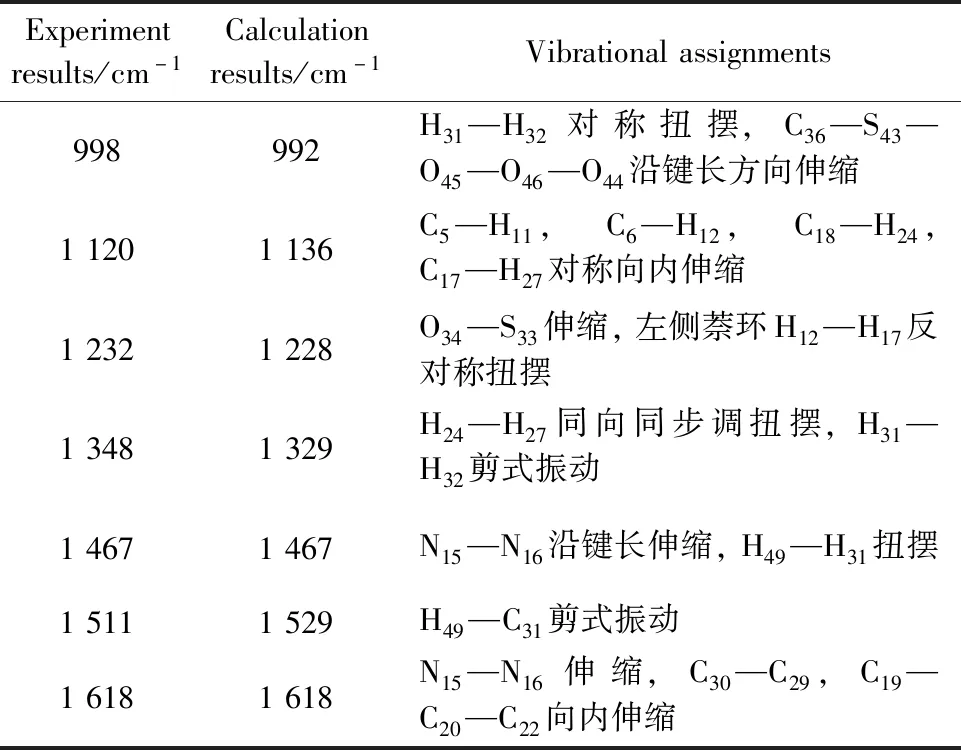
表3 苋菜红的实验拉曼特征峰与理论计算值及其归属Table 3 Experimental Raman characteristic peak and theoretical calculation value of Amaranth and its assignment
3 结 论
用显微共聚焦拉曼光谱仪对苋菜红(Amaranth)分子进行实验检测,得到苋菜红分子的实验拉曼光谱图。基于密度泛函理论,对苋菜红分子搭建分子结构进行优化,得到其稳定结构。用Gaussian 09量子化学程序包计算苋菜红分子的前线轨道、静电势及极化率,推断出苋菜红分子在偶氮基团处容易发生取代。在此基础上,对Ag1-Amaranth,Ag3-Amaranth所有的复合物在B3LYP方法下使用6-31++G(d,p)(C,H,O,N,S,Na)基组以及Sdd(Ag)基组进行结构优化和振动光谱的计算。结果说明,Ag原子对苋菜红分子起到了增强作用。最后,将实验与计算所得拉曼光谱图进行对比并归属,发现两者吻合效果较好,预测该理论的可靠性。该结果对苋菜红分子和Ag增强基底的吸附方式,解释表面增强机理具有重要意义,进而为日后用表面增强拉曼光谱法检测合成色素苋菜红提供系统严谨的理论依据。
