乌鸡中硝基咪唑类药物 检测方法的研究
2020-06-12钟雷响徐少华
钟雷响 徐少华

摘 要:本研究建立了高效液相色谱串联质谱法测定乌鸡鸡肉中羟基甲硝唑和甲硝唑含量的方法。鸡肉样品经过乙腈水溶液提取,离心取用上清液再经过PRIME HLB固相萃取柱净化、氮吹、甲醇水溶液定容以及0.22 μm水相滤膜过滤,随后用液相色谱法串联质谱法检测硝基咪唑。结果显示:在线性范围0.1 μg/L~500 μg/L内,硝基咪唑类药物线性关系良好,甲硝唑相关系数为r=0.997,羟基甲硝唑相关系数为r=0.999 3。在1.0 μg/kg、 2.0 μg/kg和10 μg/kg三个水平添加回收试验条件下,甲硝唑的回收率为80%~109%,相对标准偏差为2.45%~11.0%;羟基甲硝唑的回收率为74.0%~96.6%,相对标准偏差为3.96%~8.33%。可确定的方法检出限甲硝唑为0.35 μg/kg,羟基甲硝唑为0.38 μg/kg。该方法具有灵敏度高和重现性好的特点,可满足鸡肉中痕量元素的检测。
关键词:液相色谱法串联质谱法;硝基咪唑类药物;乌鸡;检测方法
硝基咪唑类药物是一种抗菌药物,在预防和治疗家禽的组织滴虫病和寄生虫病、牛滴虫病、猪出血性肠炎、鱼寄生虫感染和蜂微孢子虫病等疾病上有很大的作用。此类药物进入动物易感的细胞后,在无氧或少氧环境和较低的氧化还原电位下,其硝基会被电子专递蛋白还原成具有细胞毒性作用的氨基,抑制微生物细胞合成DNA,并降解DNA,破坏DNA的双螺旋结构或阻断其转录复制,从而导致微生物细胞死亡,发挥其迅速杀灭厌氧菌和有效控制细菌感染的作用[1]。体内外试验证实,硝基咪唑类药物有遗传毒性、致畸和可疑致癌的作用[2-3]。为了防止甲硝唑残留伤害人类健康,不少国家都制定了相关规定,欧盟和加拿大等地区或国家都禁止在食源性动物生产中使用甲硝唑。农业农村部在2002年颁布的227号公告《杜绝禁用兽药的滥用》中规定不准以促进动物生长为目的在食品动物的生产中使用甲硝唑及其盐和酯。本试验选取乌鸡为试验动物,一方面因为乌鸡是广东省传统的地方名菜乌鸡煲汤的主材,消费量大;另一方面由于乌鸡自身的药物代谢困难,容易造成药物在体内残留蓄积。为了保证消费者能在市场上购买到安全的乌鸡,准确测定乌鸡肉产品中硝基咪唑类药物的含量十分重要。
1 材料与方法
1.1 设备与仪器
液相色谱串联质谱仪,配有电喷雾离子源(Electrospray Ionization,ESI);Oasis? PRIME HLB(6 cc 200 mg)固相萃取小柱(美国Waters公司)、0.22 μm有机相滤膜(上海安谱公司)、电子天平(感量0.01 g、感量0.000 1 g,瑞士梅特勒公司);高速离心机(美国sigma公司);固相萃取装置;超声波清洗仪;可控温氮吹浓缩仪;涡旋振荡器。
1.2 试剂
甲硝唑和羟基甲硝唑(MNZOH)标准物质(纯度≥99%,美国DR公司),水符合GB/T 6682《分析实验室用水規格和试验方法》规定的一级水标准;甲醇(CH3OH)、乙腈(CH3CN)和甲酸(HCOOH)均为色谱纯。
1.3 试剂配制
乙腈水溶液由90%乙腈和10%水配制而成,甲醇水溶液由10%甲醇水和0.1%甲酸配制而成。
标准储备液:每种标准物质均用甲醇配制成1.00 mg/mL,在-18 ℃条件下于棕色瓶中保存。
标准工作液:根据药物的灵敏度和仪器线性范围,用甲醇水溶液配成不同浓度的混合标准工作溶液,现配现用。
1.4 乌鸡
乌鸡购自广东省深圳市各大农贸市场和商场超市。试验用的鸡肉样品取自乌鸡胸部和腿部的肌肉,经碾磨机粉碎至均匀肉泥状,装瓶保存于-18 ℃。
2 试验方法
2.1 仪器条件
2.1.1 液相色谱条件
(a)色谱柱:亚乙基桥杂化颗粒(BEH) C18(100 mm×3.0 mm,1.7 μm);(b)流速:0.3 mL/min;(c)柱温:40 ℃;(d)进样量:5.0 μL;(e)流动相及梯度洗脱程序见表1。
2.1.2 质谱参考条件
(a)离子源:电喷雾离子源(ESI);(b)扫描方式:正离子扫描/负离子扫描;(c)检测方式:多反应离子监测(Multi Reaction Monitoring Mode,MRM);(d)电喷雾电压(IS):5 500 V/-4 500 V;(e)离子源温度(TEM):550 ℃;(f)气帘气(CUR):35 psi;(g)碰撞气压力(CAD):8 psi; (h)喷雾气(GS1):55 psi;(i)辅助加热气(GS2):55 psi;(j)药物保留时间、定性定量离子对及去簇电压和碰撞电压见表2。
2.2 前处理步骤
早期研究将样品的pH调节为3,用蛋白酶过夜酶解提取与组织蛋白结合的残留物[4],但近期研究表明没必要进行酶解[5-6]。因此,本试验用90%的乙腈水溶液作为提取剂,直接提取残留药物。
2.2.1 提取
称取试样2 g(精确到0.01 g)于50 mL离心管内,加乙腈水溶液5 mL,震荡摇匀30 s,于超声波清洗仪超声5 min,用高速离心机以6 000 r/min的转速离心 10 min,转移上清液于15 mL离心管中。重复提取一次,合并上清液。上清液混匀之后再次用高速离心机以6 000 r/min的转速离心10 min。
2.2.2 净化
于上述离心后提取液中移取5 mL上清液,直接过固相萃取柱,保持1 s/滴的速度。用玻璃小管收集全部流出液,准确移出2 mL到15 mL带刻度的小管内。流出液在40 ℃水浴下氮气吹至少于0.5 mL。残留液用甲醇水溶液定容至1.00 mL,过 0.22 μm水相滤膜,待上机测定。
3 试验结果
3.1 高效液相条件确定
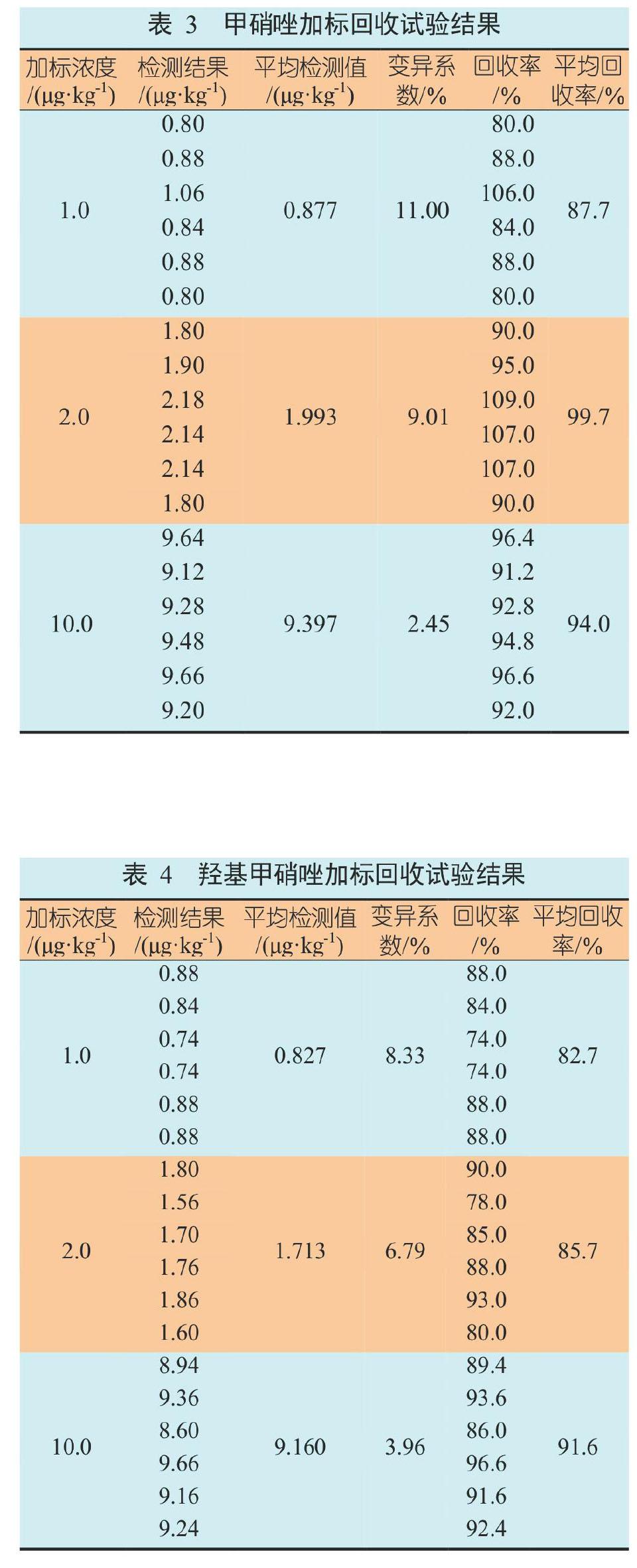
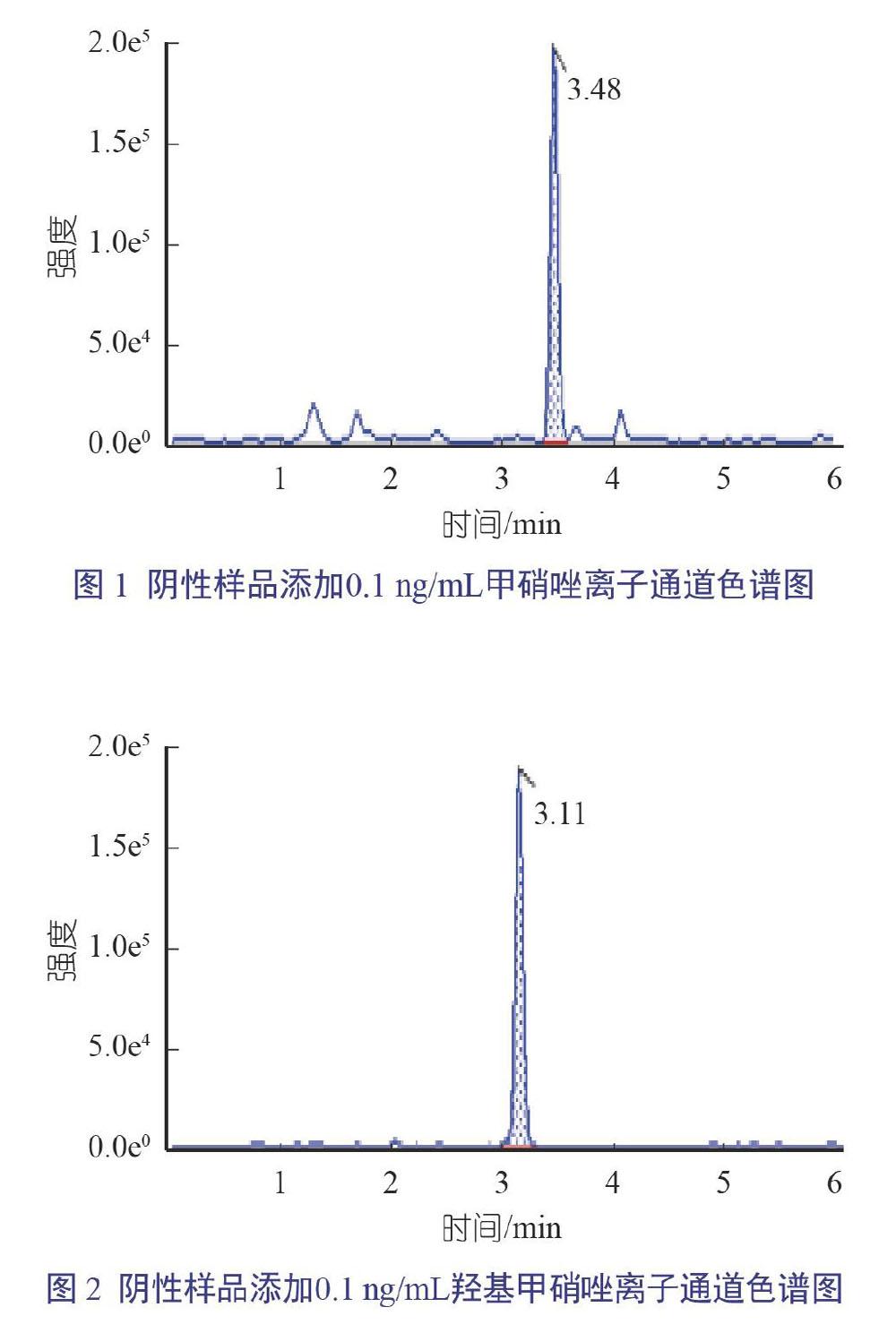
为寻找分离度好、灵敏度高且峰形尖锐的液相条件,参考相关文献及检测标准,选用ACQUITYUPLC BEH C18(1.7 μm, 3.0 mm×100 mm)色谱柱,柱温为40 ℃,采用甲酸水和甲醇作为流动相,进行梯度洗脱。进样流速为0.30 ng/mL时,硝基咪唑相应强度1.5×105,背景基线非常低,峰形尖锐,保留时间适中,可有效进行硝基咪唑的定性定量检测(图1和图2)。
3.2 前处理方法的确定
由于鸡的肌肉呈现纤维状,所以需要用本方法先对肌肉进行粉碎处理。乙腈沉淀蛋白效果较好,与水性基质互溶,是提取组织中硝基咪唑类药物的常用溶剂之一。乙腈用于提取禽肉类样品,回收率在79%~93%[7-8],故本试验选用乙腈水溶液作为提取剂。样品加入提取剂后经过超声辅助提取,可以更充分提取乌鸡肌肉中的硝基咪唑类药物。再经过高速离心,使提取剂中的残渣沉淀,其中试剂也起到了稀释的作用,有利于降低其他成分对检测仪器的信号干扰,并且可提高仪器检测的灵敏度。
3.3 线性范围,检出限检测结果
按照方法条件设置参数,配制一系列不同浓度的标准样品溶液(0.10 μg/L、 2.00 μg/L、20.0 μg/L、100 μg/L和500 μg/L),待仪器稳定后,对系列标准样品溶液进行测定,绘制其标准曲线,甲硝唑相关系数为r=0.997,羟基甲硝唑相关系数为r=0.999 3。
以乌鸡肌肉样品为基质,添加浓度为1.0 μg/kg,按照上述方法进行前处理和测定,平行样品(n)为7个,对于甲硝唑,根据各样品检测值(0.80 μg/kg、0.88 μg/kg、 1.06 μg/kg、0.84 μg/kg、 0.88 μg/kg、0.80 μg/kg和 1.02 μg/kg)計算出平均值(x)(0.897 μg/kg)及标准偏差(Sb)(0.103 6),当被分析物的回收率在70%~120%时,可计算检出限为0.35 μg/kg;对于羟基甲硝唑,根据各样品检测值(0.88 μg/kg、0.84 μg/kg、0.74 μg/kg、0.74 μg/kg、0.88 μg/kg、0.88 μg/kg和1.06 μg/kg)计算出平均值(x)(0.860 μg/kg)及标准偏差(Sb)(0.108 4),当被分析物的回收率介于70%~120%时,可计算检出限为0.38 μg/kg。计算公式如下:
3.4 方法精密度和回收试验结果
以乌鸡肌肉样品为基质,进行六组平行加标回收试验,加标浓度分别为1.0 μg/kg、2.0 μg/kg和10.0 μg/kg。将加标样品和空白样品按前述方法进行处理,上机测试,甲硝唑加标试验回收率范围分别为80%~106%、90%~109%和91.2~96.6%,羟基甲硝唑加标试验回收率范围分别为74%~88%、78%~93%和86.0~96.6%。甲硝唑及硝基甲硝唑的加标回收试验结果见表3和表4。
4 讨论
本试验讨论建立了测定乌鸡肌肉中硝基咪唑类药物含量的高相液相色谱串联质谱法。该方法通过乙腈水溶液提取、离心和氮吹等处理步骤,最终建立了甲硝唑和羟基甲硝唑的方法检出限 分别达到了0.35 μg/kg和0.38 μg/kg,线性范围为0.1 μg/L~500 μg/L。乌鸡肌肉样品在1.0 μg/kg、2.0 μg/kg和 10.0 μg/kg三个水平添加回收试验条件下,甲硝唑回收率为80%~109%,相对标准偏差为2.45%~11.00%;羟基甲硝唑回收率为74.0%~96.6%,相对标准偏差为3.96%~8.33%。该方法具有分析过程简单、准确精密和定量准确等优点,能满足乌鸡肌肉中硝基咪唑类药物的快速筛查和检测定量。
