发菜盐胁迫响应蛋白TrkA-N的克隆及在大肠杆菌中表达
2020-06-12孟广燕陈雪峰刘欢蔡国强赵圆圆党玥朱蓉静桑聪
孟广燕,陈雪峰,2*,刘欢,蔡国强,赵圆圆,党玥,朱蓉静,桑聪
1(陕西科技大学 食品与生物工程学院,陕西 西安,710021)2(陕西科技大学,陕西省农产品加工技术研究院,陕西 西安,710021) 3(西安培华学院,陕西 西安,710021)4(中国人民解放军94162部队79分队,陕西 西安,710021)
发菜,学名发状念珠藻(Nostocflagelliforme),属蓝细菌门(Cyanopyhta)、蓝细菌纲(Cyanophyceae)、断殖藻目(Hormogonales)、念珠藻科(Nostocaceae)、念珠藻属(Nostoc)[1]。发菜是陆生蓝藻的一种,藻丝体有多个形如念珠的细胞,藻丝体外包裹胶质鞘,它是原核蓝藻,生长环境恶劣,有极强生命力,能够在逆性环境下正常生长[2]。自然缺水时,发菜呈黑色细丝状,交缠成团,形如发丝,得名发菜。在全球均有发菜分布[3],它主要生长在干旱、半干旱地区的草原和荒漠及半荒漠地带,发菜在我国的分布也较广泛。它起源于古老的光能自养型低等植物,能够自给自足,有固氮特性[4],同时发菜能够耐高低温、抗干旱,有很强的自我调节能力[5]。发菜可以食用,在我国,发菜作为食物已有大约2 000年的历史,其营养丰富,富含蛋白质及锰、铁、钙等多种微量元素。焦文东[6]通过研究发现,发菜中必需氨基酸、多不饱和脂肪酸含量都较高,因此可以作为优质蛋白和二十二碳六烯酸(docosahexaenoid acid,DHA)的良好来源[7]。发菜在生长过程中会分泌活性胞外多糖[8],有很高的生物活性,能够保护细胞[9],发菜的食用和药用价值主要归功于胞外多糖的存在[10]。但也正是因此,近些年人们对其滥采乱挖,再加上发菜本身生长环境的破坏、土地盐渍化问题加剧、发菜培养不易、自然状态下生长慢等诸多因素,发菜的年产量越来越低。1999年,国务院正式批准将野生发菜列为国家二级保护野生植物,并从2000年7月起严令禁止对野生发菜的随意开采和交易[11]。
王贵春[12]通过大量研究得出结论,发菜在0.3 mol/L盐浓度下培养一段时间后比在正常条件时产生的胞外多糖质量分数增加了50.3%。赵秀霞等[13]分别测定了正常条件下培养的发菜细胞和0.3 mol /L盐浓度培养的发菜细胞的转录组序列[14],在此项研究中他们共测出 13 170 条基因的表达水平出现了一定变化[15],有些基因的表达水平上调,也有表达水平下调的基因。他们又对这些表达水平出现变化的基因进行了GO功能聚类[16]和Pathway的代谢通路分析[17],在此项研究中发现参与发菜细胞的主要盐离子运输、光合作用、部分代谢以及各种代谢产物生物合成等众多过程的基因的表达都有一定的改变[18]。本研究以在不同盐浓度下表达量变化比较明显的钾离子通道蛋白TrKA-N基因作为目的基因,从路苗等[19]的研究结果中可以得出结论,钾离子通道蛋白TrKA-N基因是影响盐胁迫下多糖分泌量的关键基因之一,通过分子生物学手段[20]克隆得到钾离子通道蛋白TrKA-N的全基因序列[21],软件推译得到该蛋白的氨基酸序列,通过各种软件分析该氨基酸编码蛋白的种类和功能,将该基因与表达载体连接后,诱导基因成功在适当条件下合成具有相应功能的蛋白质。本研究为发菜响应盐胁迫相关基因及其代谢调控机理的研究提供了一些思路,为各种蛋白基因工程菌株的成功构建[22]打下了良好基础。
1 材料与方法
1.1 材料与试剂
实验用发菜,由本实验室自行液体悬浮培养;表达载体质粒,由微生物实验室-80 ℃保藏;大肠杆菌感受态细胞及其他实验相关试剂,宝日医生物技术(北京)有限公司;专用试剂盒,天根生化科技(北京)有限公司。
1.2 仪器与设备
THZ-C-1全温振荡器,太仓市实验设备(苏州培英实验设备有限公司);MGC-400型光照培养箱,上海善志仪器设备有限公司;DYY-2C型电泳仪,北京六一仪器厂;ETC811基因扩增仪,苏州东胜仪器设备有限公司。
1.3 实验方法
1.3.1 发菜细胞培养
BG11培养基配置[23]:硅液为5.8 g/L Na2SiO3·9H2O;磷液为2.38 g/L K2HPO4·2H2O;母液A1为3.6 g/L CaCl2·2H2O;母液A2为7.5 g/L MgSO4·7H2O;母液B成分(g/L):柠檬酸,6.00;EDTA·2Na,1.00;CoCl2,0.01;CuSO4·5H2O,0.08;ZnSO4·7H2O,0.2;NaMoO4·2H2O,0.4;MnCl2·4H2O,1.8;H3BO3,2.86;FeSO4·7H2O,4.8。
培养基处理:取母液A1、A2各5 mL、母液B 500 μL于烧杯,定容到300 mL,移入锥形瓶1; Si液、P液各取5 mL,定容至200 mL,移入锥形瓶2。将瓶1和瓶2分别封口后高温灭菌(121 ℃,20 min)[24]。灭菌后把培养基放到净化工作台中,降至室温,在工作台中将瓶1和瓶2混合均匀,以200 mL/瓶的量分装至锥形瓶。发菜细胞转接量为10%,培养箱设置两段式条件循环培养,第一段为30 μmol/(m2·s)光照, 25 ℃培养12 h; 第二段为0 μmol/(m2·s)光照, 10 ℃培养12 h,培养15 d左右,可以进行后续操作。
1.3.2 目的蛋白基因TA克隆及序列测定
参照购买的植物基因组提取纯化试剂盒说明书提取发菜基因组 DNA,质量检测后于-20 ℃冰箱保藏待用[25]。
通过功能注释、代谢通路分析及美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)数据库比对,筛选出钾离子通道蛋白TrKA-N基因,同源比对发现该基因与点状念珠藻钾离子通道蛋白TrKA-N基因的相似度为92.41%。根据转录组测序结果与NCBI相似基因序列同源比对分析后设计引物,并在上下游引物分别加入BamH I和XhoI的酶切位点,最终设计引物为上游引物ATF:5′-CGCGGATCCGTGAATCTTTCATCATTAAGT-3′,下游引物ATR:5′-CCGCTCGAGAATCCGGCAAACGATTGATAT-3′,引物由上海捷瑞生物工程有限公司合成。20 μL PCR 反应体系为发菜DNA 2 μL,设计合成的上下游引物各1 μL,2×Taq PCR Master Mix 10 μL,超纯水6 μL。PCR条件为95 ℃反应5 min,95 ℃反应30 s,50 ℃,30 s,72 ℃反应2 min,重复反应30次,72 ℃反应5 min。反应完成后,电泳检测纯化后产物与pMD19-T载体重组。将重组基因转化到大肠杆菌DH5α 感受态细胞,菌液进行PCR验证成功后,扩大培养,抽提质粒,质粒送至北京奥科鼎盛生物科技公司测序。
1.3.3 序列分析
将测序序列与NCBI数据库中序列进行BLAST比对,确认基因信息,用DNAMAN推译出相应氨基酸序列,再用DNAstar和ProtParam进行分析,利用TMHMM分析目标蛋白质的跨膜区,用ProtScale分析该蛋白的亲水性及疏水性,分别用DNAMAN 和SOPMA分析预测该蛋白的二级结构,用NetPhos3.1 Server分析该蛋白的磷酸化位点及个数。
1.3.4 TrkA-N钾通道蛋白基因原核表达
参考测序结果,重新设计引物,并且在上下游引物的 5′端分别添加BamH I 和XhoI的酶切位点及保护碱基,最终设计引物为上游引物ATF1:5′-GGACAGCAAATGGGTCGCGGATCCGTGAATCTTTCATCA-TTAAGT-3′,下游引物ATR1:5′-CGACGGAGCTCGA-ATTCGGATCCAATCGGCAAACGATTGATAT-3′,合成引物后,以重组质粒为模板,PCR扩增体系与前面实验一致,退火温度改为52 ℃,其他条件不变。扩增完成后检测并纯化产物,产物和pET28a质粒分别用2种酶BamH I 和XhoI做双酶切反应,然后连接2个片段,连接成功的重组基因就是蛋白基因表达载体。将重组质粒转化到大肠杆菌DH5α培养,挑选阳性克隆进行菌液PCR及双酶切验证,转化大肠杆菌BL21(DE3),诱导表达进行聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,SDS-PAGE)。
2 结果与分析
2.1 目的蛋白基因TA克隆
抽提发菜DNA,电泳30 min,结果如图1-a所示,提取的发菜DNA条带清晰,没有杂带,说明其质量较好。目的基因体外扩增反应的电泳结果如图1-b所示,在预期大小的分子质量标准附近(700 bp)有特异条带,说明实验正确。TrkA-N钾通道蛋白基因连接pMD19-T载体并转化感受态细胞后的菌液PCR电泳结果如图1-c所示,克隆位点大小是140 bp,加上目的基因700 bp左右,在800 bp左右出现特异清晰条带,说明结果正确,即发菜的TrkA-N钾通道蛋白基因与pMD19-T载体连接成功,可以进行后续实验。
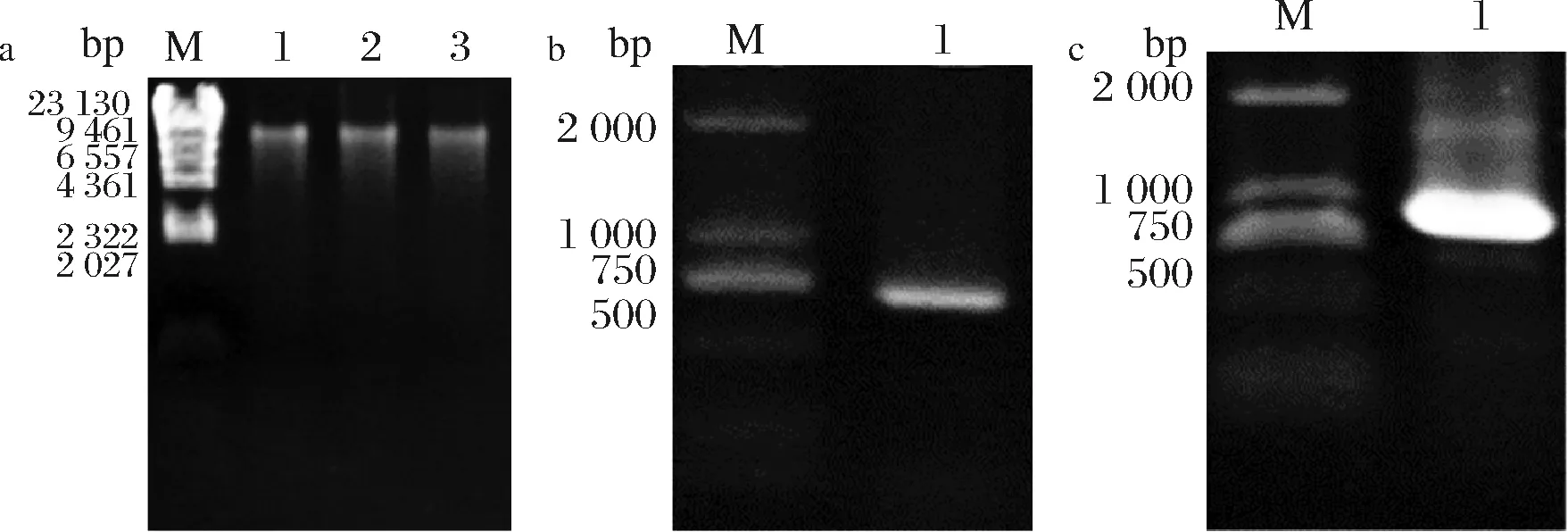
a-发菜DNA电泳图;b-锂离子通道蛋白TrkA-N基因的PCR扩增;c-钾离子通道蛋白TrkA-N基因重组质粒阳性转化(DH5a/AT)PCR验证图1-a:泳道M-分子质量标准λDNA HindⅢ,泳道1~3-发菜DNA;图1-b:泳道M-分子质量标准D2000,泳道1-目的基因;图1-c:泳道M-分子质量标准D2000,泳道1-成功重连的产物条带图1 发菜DNA提取、目的基因体外扩增及重组质粒阳性转化验证Fig.1 DNA extraction, PCR amplification of the target gene in vitro and positive transformation of recombinant plasmids were verified
2.2 目的蛋白核酸及氨基酸序列分析
2.2.1 核苷酸及氨基酸序列
钾离子通道蛋白TrkA-N基因与pMD19-T载体成功连接后,用pMD19-T载体通用引物对其双向测序,结果如图2所示,发菜钾离子通道蛋白TrkA-N基因片段大小为696 bp,推译得到的氨基酸序列表明,该蛋白共有231个氨基酸,DNAstar分析可知,带电荷的氨基酸有55个,占总氨基酸残基个数的23.81%,其中25个带正电荷,30个带负电荷。极性氨基酸有53个,占22.94%。疏水性氨基酸92个,占39.83%,酸性氨基酸和碱性氨基酸分别占13.42%和10.82%。在钾离子通道蛋白TrkA-N中包含较多的氨基酸为 Leu(10.8%)、Val(10.0%)、Ala (7.8%)和Gly(6.9%),其不稳定值为34.74(<40),说明钾离子通道蛋白TrkA-N为稳定蛋白。

图2 钾离子通道蛋白TrkA-N基因核苷酸及氨基酸序列Fig.2 Nucleic acid and amino acid sequence of potassium channel protein TrkA-N gene
用DNAMAN把目的蛋白的核苷酸序列和它对应的氨基酸(amino acid,AA)序列与NCBI数据库里的同源物种比对,结果如图3所示,通过分析比对发现,发菜钾离子通道蛋白TrkA-N基因保守性较高,核苷酸序列与念珠藻(Nostocsp.′Peltigera membranacea cyanobiont N6′)的相似度为93.25%,与点状念珠藻(NostocpunctiformePCC73102)的相似度为92.41%,与念珠藻(NostoclinckiaNIES-25)的相似度为90.12%,与筒孢藻(Cylindrospermumsp. NIES-4074)的相似度为85.42%,与丝状蓝藻(Calothrixsp. NIES-2098)的相似度为84.23%,(Anabaenasp. WA102)的相似度为84.02%,与丝状蓝藻(Calothrixsp. PCC 7507)的相似度为83.59%。
目的蛋白的AA序列在进化过程中的变化很小(图4),通过分析比对发现,发菜钾离子通道蛋白TrkA-N序列具有较高的保守性,AA序列与点状念珠藻(NostocpunctiformePCC73102)的相似度为96.86%,与念珠藻(Nostocsp. KVJ20)的相似度为96.32%,与念珠藻(Nostoclinckia)的相似度为94.12%,与念珠藻(Nostoccalcicola)的相似度为93.32%,与念珠藻(Nostocsp. T09)的相似度为91.78%,与眉藻(Calothrixsp. PCC 7507)的相似度为91.12%,与柱孢藻(Cylindrospermumstagnale)的相似度为90.31%,与鱼腥藻(Anabaenasp. 90)的相似度为89.67%,与眉藻(Calothrixsp. NIES-2098)的相似度为90.12%,与鱼腥藻(Anabaenasp. AL09)的相似度为89.23%。

图3 钾离子通道蛋白TrkA-N核苷酸序列进化树Fig.3 Phylogenetic tree of ptassium channel protein TrkA-N nucleotide sequence
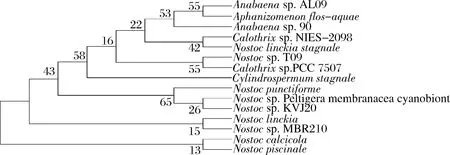
图4 钾离子通道蛋白TrkA-N氨基酸序列进化树Fig.4 Phylogenetic tree of ptassium channel protein TrkA-N amino acid sequence
2.2.2 钾离子通道蛋白TrkA-N基因分析及性能预测
利用软件ExPASy检测目的蛋白的疏水性,结果如图5所示,第15位精氨酸(Arg)亲水性最强,第85位异亮氨酸(Ile)疏水性最强,该蛋白的亲水性区间多于疏水性区间,因此确定钾离子通道蛋白TrkA-N为亲水性蛋白。

图5 钾离子通道蛋白TrkA-N疏水性分析Fig.5 Hydrophobicity analysis of potassium channel protein TrkA-N
利用 TMHMM 程序对目的蛋白进行跨膜区分析,根据数据分析显示,该蛋白不含跨膜区。
利用NetPhos3.1 Server分析目的蛋白的磷酸化位点及位点个数,结果显示,目的蛋白氨基酸序列中磷酸化位点个数分别为酪氨酸2个,苏氨酸6个,丝氨酸10个。
通过2个软件DNAMAN和SOPMA分析预测目的蛋白的二级结构,虽然2个软件分析的方式不同,给出结果的形式不同,但是最终得出的结论一致(表1),即蛋白的二级结构主要为β折叠和α螺旋。

表1 发菜钾离子通道蛋白TrkA-N二级结构预测结果Table 1 Prediction results of TrkA-N secondary structure of potassium channel protein in Nostoc flagelliforme
2.3 钾离子通道蛋白TrkA-N在大肠杆菌中的表达
构建目的蛋白基因的表达载体pET28a-TrkA-N potassium channel protein gene,然后转化 DH5α 挑选阳性克隆,在液体培养基扩大培养后进行菌液PCR验证,得到1 000 bp左右的特异条带(加上克隆位点300 bp左右),如图6所示。抽取质粒后进行双酶切验证反应,电泳后可以看到两条比较清晰的条带,大小分别在700 bp和5 000 bp左右,与预期大小一致(图7)。测序结果表明插入片段就是钾离子通道蛋白TrkA-N基因。

泳道M-分子质量标准D2000;泳道1-钾离子通道蛋白TrkA-N基因重连产物图6 目的蛋白基因重组质粒阳性转化的PCR验证Fig.6 PCR identification of positive transformant

泳道M-分子质量标尺D15 000+2 000;泳道1-目的蛋白基因的双酶切图7 钾离子通道蛋白TrkA-N基因重组质粒阳性转化子的酶切验证Fig.7 Enzymatic digestion of recombinant plasmid positive inverters of potassium channel protein TrkA-N gene
将表达载体转化到大肠杆菌BL21细胞,挑选阳性菌接种到含有卡那霉素的 LB 培养基,37 ℃,200 r/min培养,10 h后每隔1 h测定1次菌液OD600值,OD值达到0.8时,加入诱导剂异丙基-β-D-硫代半乳糖苷(isoproyl-β-D-1-thiogalactopyranoside,IPTG),保证其终浓度为1 mmol/L,对照组分别为加了IPTG且终浓度为1 mmol/L的pET-28a空质粒菌和没有加IPTG的阳性菌,37 ℃,120 r/min培养20 h后取样进行SDS-PAGE电泳,结果如图8所示,表明有我们预期的外源蛋白表达,目的蛋白分子质量为31.41 kDa,外源蛋白分子质量与预测的目的蛋白分子质量相符,说明钾离子通道蛋白TrkA-N表达成功。
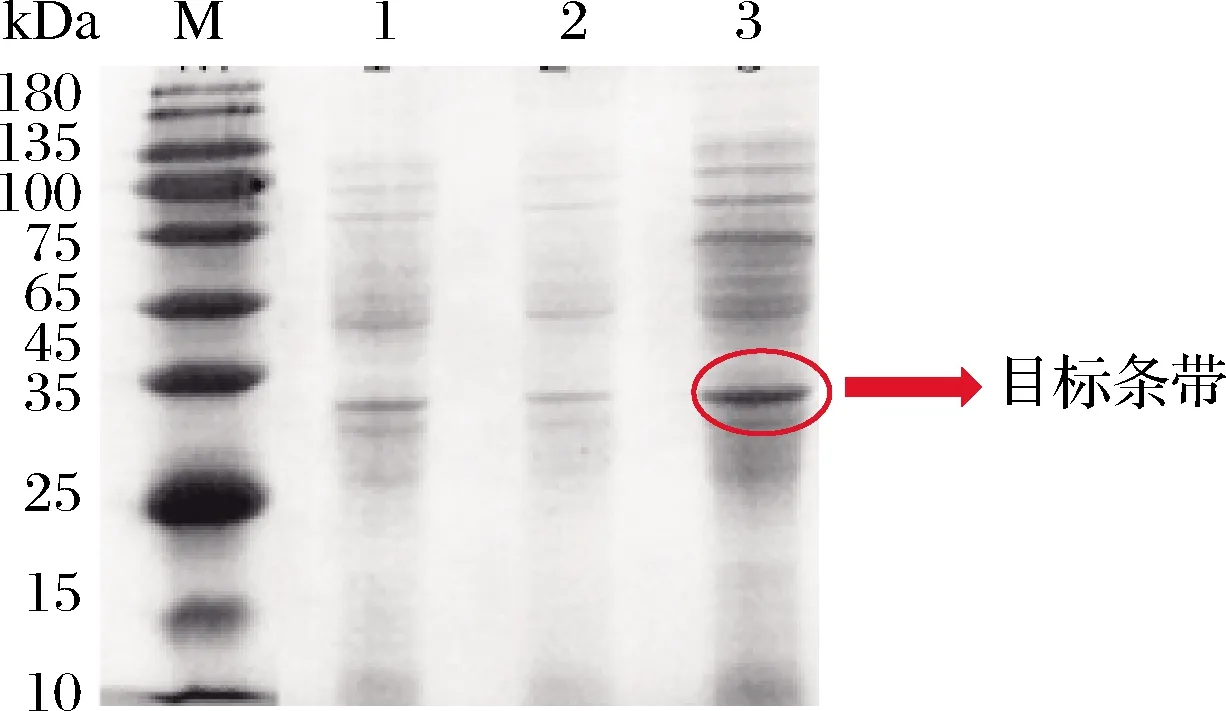
泳道M-基因表达验证参考10~180 kDa;泳道1-1 mmol /LIPTG诱导20 h的空白质粒;泳道2-不加诱导剂的阳性质粒转化子;泳道3-1 mmol /L IPTG诱导20 h的目标质粒图8 钾离子通道蛋白TrkA-N基因在大肠杆菌BL21中的表达Fig.8 Expression of TrkA-N potassium channel protein in E.coli BL21
3 结论与讨论
随着土地盐渍化问题的加剧,盐胁迫问题已经成为了一个非常常见的非生物胁迫,在一定浓度的盐胁迫作用下,发菜细胞的胞外多糖产量增加,其内在因素是发菜中与盐胁迫相关基因的转录水平发生了一些不同程度的改变,一部分基因转录水平上调,另一部分基因表达水平下降,其中钾离子通道蛋白TrkA-N基因的转录水平出现了较明显的下调,说明该基因可能是参与一定盐浓度作用下胞外多糖代谢过程重要的基因之一,可能在发菜盐胁迫作用下的一些相关反应中发挥了比较重要的作用。钾离子通道蛋白TrkA-N基因可以参与多糖合成,也验证了发菜可以通过改变一些基因的转录水平对盐胁迫做出相应缓解作用的反应,该结果从分子水平解释了盐胁迫下发菜多糖产量提高的调控机理,为该结论提供了有力的依据。本研究利用分子生物学方法体外扩增钾离子通道蛋白TrkA-N编码基因,与pET28a质粒重连构建表达载体并转化大肠杆菌BL21(DE3)诱导表达,最佳表达条件为液体培养10 h后待菌液OD600值达到0.8时,添加终浓度为1 mmol/L诱导剂 IPTG,继续诱导20 h,最终在大肠杆菌中成功表达出分子质量为31.41 kDa的蛋白,本实验在成功构建表达载体的基础上进一步摸索出诱导目的基因在该载体中成功表达的最优条件,为更多基因的研究提供了较成熟的研究思路,也为更深入的研究打下了良好基础。然而,正如前文所说,参与盐胁迫反应的基因还有很多,在该作用下多糖产量变化的过程和机制也相当复杂,因此,要研究的基因以及这些基因编码蛋白质的种类及功能还有很多,它们在盐胁迫下的响应机理以及调控多糖代谢机制还需进一步研究。
