多基因模型在肝细胞癌预后中的应用
2020-06-09魏之菡法博涛俞章盛
魏之菡 法博涛 俞章盛
(上海交通大学生命科学技术学院生物信息学与生物统计学系 上海交通大学-耶鲁大学联合生物统计与数据科学中心,上海 200240)
肝癌是全球恶性肿瘤患者死亡的主要原因之一,在我国2017 年肿瘤死亡排名中高居第二位,而肝细胞癌(Hepatocellular carcinoma,HCC)在原发性肝癌中占比高达70%-90%[1-3]。近年来,HCC 的治疗措施有了很大的改善,包括手术切除、射频消融术、肝移植、栓塞化疗术、靶向药物治疗等,但是HCC患者的5 年生存率仍然偏低[4-5]。另一方面,处于同一肿瘤分期的HCC 患者可能有不同的预后表现,故仅基于临床指标来预测HCC 患者预后可能不尽人意[6]。因此,引入分子水平信息对患者进行更加精准的风险分型十分必要,将促进精准医疗的开展。
目前,利用分子标志物进行HCC 风险分型预测的模型的大部分研究利用基因差异表达分析筛选出的mRNA 或miRNA 建立风险打分模型进行HCC 患者风险预测,每次通过中位数或软件设置单数据集打分分界点将HCC 患者分成高低风险组来验证模型的准确性,但用这些模型进行分型难以保证不同数据集的打分分界点的统一性[7-12]。Liu 等[13]整合多组学数据将HCC 分成5 种类型,部分类型却呈现类似的总体生存期表现。有些模型虽然稳健,但需要较多的分子信息来进行模型构建,如Chaudhary 等[2]用深度学习的方法整合mRNA、miRNA 及甲基化等多组学数据,Fa 等[14]基于多条通路或通路子集将HCC 分成两类生存风险有差异的组别。因此,如何利用较少的分子信息来建立预后模型,既能保证较高的预测稳健性与准确性,又具备操作简单、诊断成本低廉的特点,是本研究的主要目标。
本研究首先针对HCC 预后相关的多条通路,通过主成分分析筛选出41 个核心基因。然后通过k 均值聚类、支持向量机等机器学习的方法在TCGA 数据集上建立HCC 患者分组预后模型,并在TCGA 数据集内部及3 个外部数据集上进行稳健性、有效性验证。最后通过HCC 患者组间基因差异表达分析和富集分析,筛选出与HCC 疾病进展有关的基因与通路,以期为个性化医疗提供理论依据,同时进一步验证模型分类的有效性。
1 材料与方法
1.1 材料
肝细胞癌患者基因表达和临床数据来自4 个数 据 集TCGA、LIRI-JP、GSE14520 及GSE54236。其中,TCGA 数据集通过TCGA-Assembler 2.0.6 软件从癌症和肿瘤基因图谱(The Cancer Genome Atlas,TCGA)数 据库下载(https://tcga-data.nci.nih.gov/tcga/)[15];LIRI-JP 数据集来自国际肿瘤基因组协作组(International Caner Genome Consortium,ICGC) 数 据 库(https://dcc.icgc.org/projects/LIRIJP)[16];GSE14520 和GSE54236 两个 数 据集 来 自NCBI 的GEO 数 据 库(https://www.ncbi.nlm.nih.gov/geo/)[17-18]。去除生存时间不正确或缺失及非HCC 患者数据,最终得到不同样本量的HCC 患者数据集如表1 所示。其中,数据集GSE54236 缺乏生存时间删失(Censoring)信息。

表1 HCC 数据集介绍
1.2 方法
1.2.1 基于信号通路的基因筛选及HCC 患者预后分组模型的建立 根据文献中提到的与肝细胞癌生存风险显著相关的13 个信号通路或通路子集[14],我们分别对TCGA、LIRI-JP 及GSE14520 三个HCC 数据集相应信号通路中的基因表达数据进行主成分分析(Principle component analysis,PCA),分别截取主成分载荷(Loading)大于0.3 及总方差的累积解释比例(Cumulative proportion)大于0.8 的主成分后取交集,得到通路中的关键基因。PCA 分析使用R语言内置的prcomp 函数。
基于上一步得到的与生存相关的关键基因,首先对整个TCGA数据集做无监督K均值聚类(K-means clustering,k=2),将HCC 患者分为两组。用logrank 检验(R 包 survival[19])比较两组间生存风险差异,将高风险组记为S1(Subgroup 1),低风险组记为S2(Subgroup 2)。然后使用支持向量机(Support vector machine,SVM)进行有监督建模。模型构建与验证采用R 语言内置的kmeans 函数和R 包e1071中svm 函数(https://cran.r-project.org/web/packages/e1071/index.html),其中SVM 模型的最优参数使用交叉验证的方法选择。
为了验证该模型的稳定性,我们用整个TCGA数据集进行交叉验证,即将数据集随机分成5 个大小一致的组,任意选择3 组做训练集,其余2 组做测试集,共得到10 个数据组合进行验证。为了评价模型的预测能力,我们将整个TCGA 数据集作为训练集,验证模型在LIRI-JP、GSE14520 及GSE54236数据集中的预测能力。
对组别间生存风险差异评价使用以下3 种衡量指标:一致性指数(Concordance index,C-index,R包survcomp[20])、log-rank 检 验 的P 值(Log-rank P value of Cox-PH model)及Brier 分数(Brier score,R包survcomp)。一致性指数由Harrell 提出[21],该值范围在0 到1 之间,越大表示模型预测结果与实际生存风险排序越一致。总生存期(Overall survival,OS)的KM(Kaplan-Meier)曲 线 绘 制 使 用R 包survminer 中的ggsurvplot 函数(https://cran.r-project.org/web/packages/survminer/index.html),并用log-rank检验比较组间生存差异。Brier 分数衡量预测结果与真实结果间的差异,该值越小表示模型预测能力 越好。
在建模之前,对训练集及测试集进行了两步归一化处理。首先,对所有数据集分别进行中位数标准化,即利用该数据集所有肿瘤样本对应基因表达值的中位数(Median)和绝对中位差(Median absolute deviation,MAD)对该基因表达值进行标准化。其次,利用TCGA 训练集的各基因归一化后的均值及方差对测试集数据进一步标准化。需要指出的,无论是测序数据还是芯片数据,我们均用各数据内最小值(近似仪器检测下限)来填补缺失值,未log2 处理的数据进行对数化。针对芯片数据多个探针对应一个基因的情况,我们取其平均值。
1.2.2 基于单因素Cox 回归的基因筛选及HCC 患者预后分组模型的建立 为了与上述基于信号通路中关键基因的模型做对比,利用单因素Cox 回归进行基因筛选。取TCGA、LIRI-JP 及GSE14520 3 个HCC 数据集的共有基因,然后在TCGA 数据集上,利用单因素Cox 回归筛选上述基因中与生存风险最显著相关的子集(Log-rank P 值最小)。为保证可比性,筛选的基因数目、HCC 患者预后分组模型的建立方法及稳定性、预测能力的检验与1.2.1 相同。
1.2.3 HCC 疾病进展相关的差异基因筛选 应用
1.2.1 中构建的风险预测模型可分别将3 个主要数据集TCGA、LIRI-JP 及GSE14520 中的HCC 患者分成两个生存风险存在显著差异的组别。我们对两组间的肿瘤样本(HCC 高风险与低风险组相比)进行差异表达基因(Differentially expressed genes,DEGs)筛选,其中测序数据、芯片数据分别使用R 包DESeq2 和limma 进行分析[22-23]。差异检验使用BH(Benjamini-Hochberg)方法控制假阳性率[24]。对3 个数据集中满足|log2FoldChange|>1、校正后P 值<0.05 的差异表达基因取交集进行后续分析。
1.2.4 差异表达基因的功能富集及注释 使用R 包clusterProfiler 对上一步得到的与HCC 疾病进展有关的差异基因进行基因本体(Gene ontology,GO)富集分析和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)信号通路分析,探索与HCC 疾病进展有关的生物学过程[25]。筛选条件均为BH 校正后的P 值<0.01,q 值<0.05。
1.2.5 差异表达基因的蛋白质互相作用网络及子网络构建 使用STRING 11.0 数据库(https://stringdb.org)分析与HCC 疾病进展有关的差异表达基因的蛋白质相互作用网络(Protein-protein interaction network,PPI),并用Cytoscape 软件进行可视化处理[26-27]。其中蛋白质节点的度(Degree)用插件CentiScaPe 计算,图示节点大小与度成正比,并用插件MCODE(Molecular complex detection)分析子网络[28-29]。对度排名前20 的蛋白质,与KEGG 通路过程(P 值<0.05)取交集,得到hub 基因(Hub genes)。使 用 插 件ClueGo+CluePedia 分 析hub 基因参与的重要KEGG 通路过程[30-31]。
1.2.6hub基因的预后相关性分析 将TCGA 数据集中355 例HCC 患者的总体生存时间与hub基因表达特征进行统计检验,探索这些hub基因对肝细胞癌的预后价值。根据每个hub基因表达水平的中位数将HCC 患者分为高表达组(表达值>中位数)和低表达组(表达值≤中位数),通过KM 曲线图、logrank 检验来研究两组之间的总生存期差异。
2 结果
2.1 基于信号通路的基因筛选结果及HCC患者预后分组模型的表现
根据文献中提到的与HCC 生存风险显著相关的13 条信号通路或通路子集,我们分别对TCGA、LIRI-JP 及GSE14520 三个HCC 数据集中相应信号通路中的基因表达数据进行主成分分析,取交集后得到41 个核心基因:CDK1、DUT、EGF、ENO1、FOXM1、G6PD、GMDS、GNAS、GNG4、GPI、GPX7、HEXA、IRAK1、ITPR1、PPP2R5A、MAPK1、MAPK9、MAPK13、MAP2K2、RAC1、RAD51、RAF1、RENBP、RRM2、TGFA、TK1、TKT、TYMS、UGDH、UCK2、IKBKG、SQSTM1、WASF1、GNPDA1、GNB5、RALBP1、PLCB1、CYB5R1、HKDC1、NPL和UAP1L1。
根据信号通路筛选的41 个基因及无监督聚类得到的标签对TCGA 基因表达数据建立HCC 风险预测模型,其中稳健性及准确性分析结果如表2 所示。TCGA 数据集交叉验证结果表明,模型的预测准确率在0.71 附近,HCC 患者组间总体生存期的logrank 检验P 值平均为0.03,且Brier 分数较低。在其他3 个数据集上,该模型区分的HCC 患者组间总体生存期均存在显著差异(log-rank 检验P 值<0.05),预测准确率均高于0.7(一致性指数>0.7)。4 个数据集的KM 曲线(图1)可进一步直观表明,该模型可将HCC 患者分成生存风险存在显著差异的两组(S1 高风险组和S2 低风险组)。其中,TCGA 数据集log-rank 检验P 值为0.000 18,LIRI-JP 数据集log-rank 检验P 值为0.000 38,GSE14520 数据集logrank 检验P 值为0.002 1,GSE54236 数据集log-rank 检验P 值为0.012。
2.2 基于单因素Cox回归的基因筛选结果及HCC患者预后分组模型的表现
针对TCGA、LIRI-JP 及GSE14520 三个HCC 数据集的共有基因,利用单因素Cox 回归得到TCGA数据集中与生存风险最显著相关的41 个基因:CDCA8、SFPQ、KIF20A、G6PD、PSRC1、UTP11、TTC26、VRK2、KIF2C、CENPA、NEIL3、CCNJL、UCK2、TTK、ZNF643、NCAPG、YBX1、NDC80、PDE6A、TXNRD1、TPX2、SEPHS1、HDAC2、DYNC1LI1、DBF4、HJURP、DLGAP5、CHORDC1、C19orf26、CEP85、MCM10、CPSF6、EZH2、CBX2、GLMN、NCAPD2、GNL2、ZNF239、NUP205、KIF18A及TRIP13。
根据单因素Cox 回归筛选的41 个基因及无监督聚类得到的标签对TCGA 基因表达数据再次建立HCC 风险预测模型,其中稳健性及准确性分析结果如表2 所示。TCGA 数据集交叉验证结果表明,该模型的预测准确率也在0.71 附近,HCC 患者组间总体生存期的log-rank 检验P 值平均为0.02,且Brier分数较低。在其他3 个数据集上,该模型区分的HCC 患者组间总体生存期均存在显著差异(log-rank检验P 值<0.05),预测准确率均高于0.65(一致性指数>0.65)。

表2 两个模型在TCGA 数据集做交叉验证及外部数据上的预测表现
2.3 HCC疾病进展相关的基因差异表达分析与富集分析的结果
基于信号通路中41 个核心基因的模型可将HCC 患者分成高风险组S1 和低风险组S2,S1 组患者的总体生存时间显著低于S2 组。通过对TCGA、LIRI-JP 及GSE14520 三个HCC 数据集各自S1 与S2组之间进行基因差异表达分析,最终筛选出61 个上调基因及122 个下调基因。3 个数据集所得的差异表达基因数目具体如图2-A 所示。
对上述得到的HCC 疾病进展相关的差异表达基因进行GO 富集分析(前10 个显著结果见图2-B),发现上调基因主要富集在染色体分离、细胞核分裂、细胞器分裂、细胞核有丝分裂等生物学过程,下调基因主要富集在小分子分解代谢过程、类固醇代谢过程、羧酸生物合成过程等生物学代谢过程。对差异表达基因进一步进行KEGG 通路富集分析(图2-C),发现上调基因主要涉及细胞周期、卵母细胞减数分裂、p53 信号通路、DNA 复制等信号通路,下调基因主要涉及视黄醇代谢、细胞色素P450 蛋白负责的药物代谢等代谢通路。

图1 四个数据集的两组间总体生存期差异
2.4 差异表达基因的蛋白质互作网络及hub基因 筛选
利用STRING 11.0 数据库与Cytoscape 3.7.2 软件,构建如图3 所示的162 个节点、1 172 条边的差异表达基因的蛋白质互作网络。通过插件MCODE分析后发现两个主要的子网络模块1(35 个节点,557 条边,Score=34.353)和模块2(28 个节点,174条边,Score=12.889),图示环状。将编码度排名前20 的结点蛋白质的差异基因与KEGG 通路富集过程取交集,得到12 个hub基因:CCNB1、CDK1、
RRM2、BUB1B、CCNB2、TTK、CDC20、MCM4、RFC4、PTTG1、MCM2和MAD2L1。这12 个基因均属于模块1 的相互作用网络,包含66 条边的互作关系(图4-A),经过KEGG 富集分析发现主要集中在细胞周期、卵母细胞减数分裂等信号通路(图4-B)。
2.5 hub基因的预后分析结果
根据TCGA 数据集中的355 例HCC 患者的生存数据,对12 个hub基因分别进行预后差异分析。如图5 所示,除了基因MCM4,每个基因高表达组的生存风险率均显著高于低表达组(Log-rank P 值<0.05)。
3 讨论
肝细胞癌是全球常见的恶性肿瘤之一,呈现出发病机制复杂、风险因素多样的特点[4]。因此,融入分子水平信息对患者进行准确分类十分关键,能够指导不同分型患者的个性化治疗方案设计。

图2 差异表达基因(S1 和S2 组相比)的筛选及功能注释

图3 差异表达基因的蛋白质相互作用网络分析(组S1 和S2 相比)
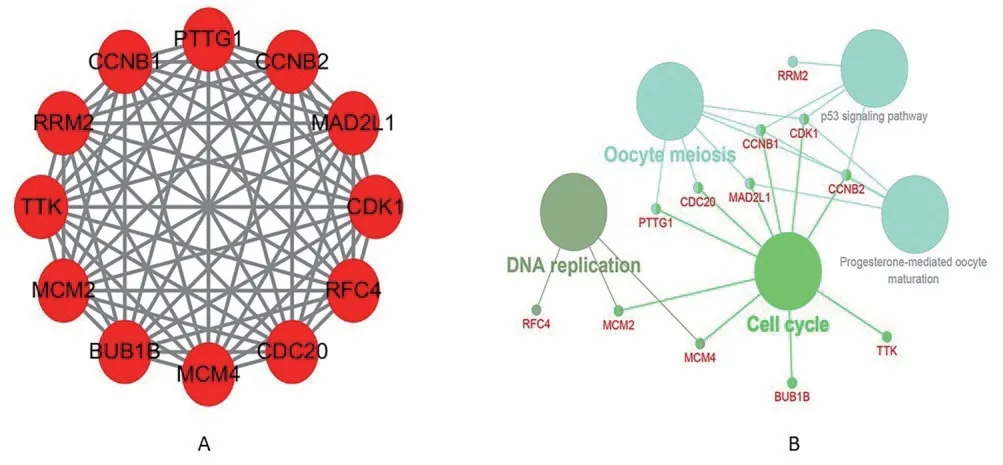
图4 hub 基因的分析研究
本研究利用信号通路中41 个核心基因的表达数据,通过k 均值聚类、支持向量机等方法在TCGA数据集上建立了HCC 分类模型。该模型能够将HCC患者分成生存风险存在显著差异的两组,交叉验证结果表明模型具有较好的稳健性,并且3 个外部数据集的测试结果证明该模型能够对HCC 患者进行准确分类。基于信号通路中41 个核心基因的模型与基于单因素Cox 回归筛选41 个基因的模型在TCGA 数据集上交叉验证和LIRI-JP 数据集上准确性验证的结果差别不大,甚至从某种程度上来说(log-rank 检验P 值),基于单因素Cox 回归的模型表现更好。但是从GSE14520 和GSE54236 两个数据集上的表现来看,基于信号通路核心基因的模型预测准确性(一致性指数>0.7)远好于基于单因素Cox 回归的模型(一致性指数>0.65)。综上所述,这两个模型均能对患者的生存风险进行有效区分,但是基于信号通路中核心基因的模型在多个数据集上的整体表现更好,更稳定并准确预测HCC 患者的生存风险。本研究的工作也是围绕信号通路中核心基因的模型而展开。与基于深度学习方法进行HCC 预后预测的文献结果相比,模型在3 个相似的数据集上有不逊色的表现,而且需要的基因数更少,无需甲基化、miRNA 的数据,能够进一步降低患者检测成本[2]。
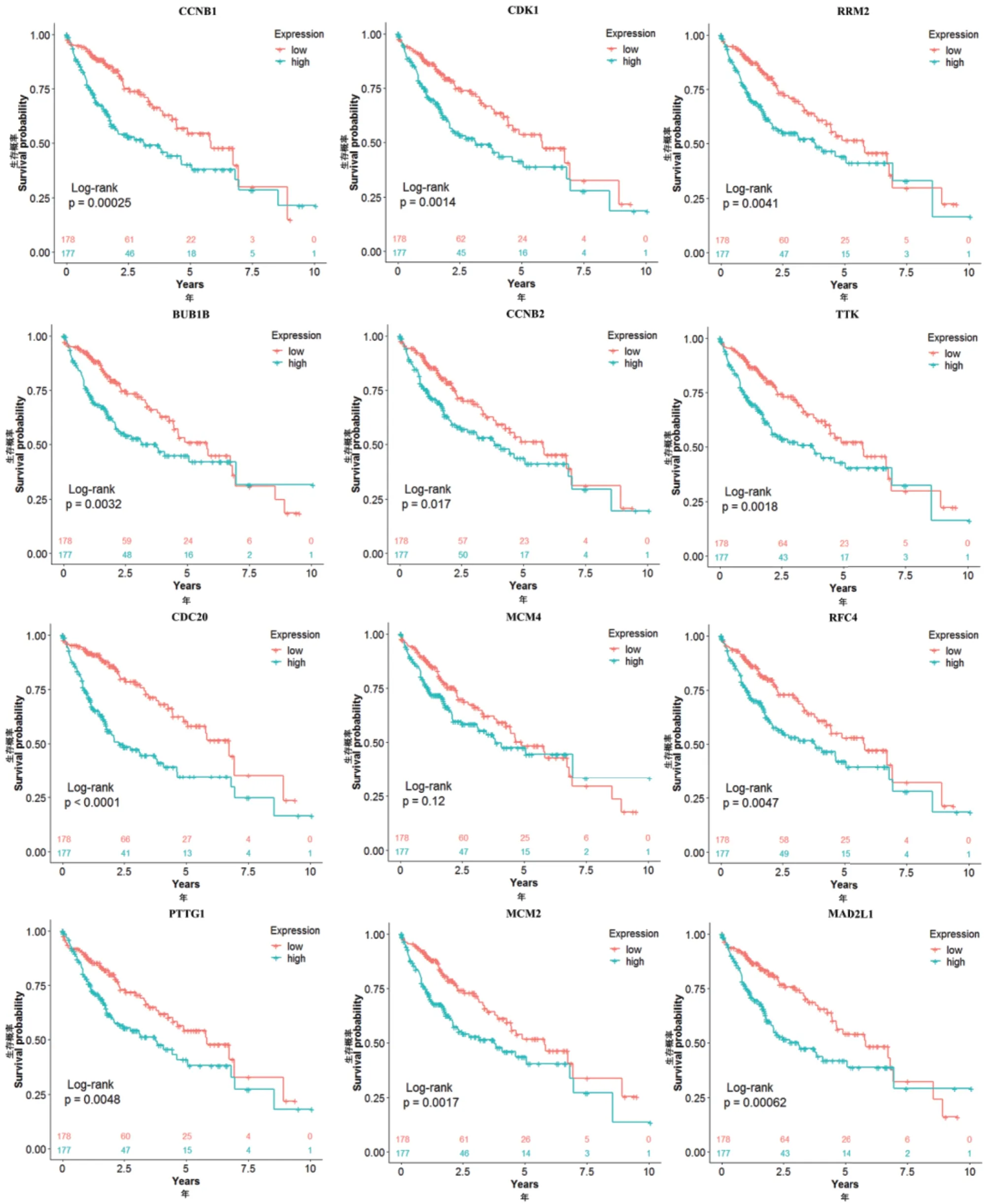
图5 12 个hub 基因在肝细胞癌中的预后价值(总体生存期)
对分类后生存风险差异显著的两组HCC 患者的表达数据进行生物信息学分析,发现高风险组的上调基因主要富集在染色体分离、细胞核分裂、细胞器分裂、细胞核有丝分裂等生物学过程(GO 分析)及细胞周期、卵母细胞减数分裂、p53 信号通路、DNA 复制等信号通路(KEGG 分析)。其中染色体分离、细胞核分裂、细胞器分裂、细胞核有丝分裂、卵母细胞减数分裂及DNA 复制等都是发生在细胞周期中的生物学过程,p53 信号通路最终也诱导细胞周期的调控,而细胞周期调控和癌症进展关系紧密,细胞周期失调在促进肝细胞癌发生中起核心 作用[32-33]。
本研究共发现12 个hub基因,除基因MCM4外,每个基因高表达组的生存风险率均显著高于低表达组,而它们在S1 组中的相对高表达或许部分解释了S1 组与S2 组HCC 患者间的生存风险差异。这些基 因CCNB1[4,34-37]、CCNB2[35-37]、CDK1[34-37]、R R M 2[35-37]、B U B 1 B[34,36-37]、T T K[35,37]、CDC20[4,34,36-37]、MCM2[36]、MCM4[36]、RFC4[36]、PTTG1[35-36]和MAD2L1[4,34,36-37]均 在HCC 癌 组织中表达显著上调,该结果进一步表明分类模型的有效性,并说明这些基因可能在HCC 的发生、进展中起关键性作用,可以作为疾病预警的重要标志物或者潜在治疗靶点。要说明的是,MCM2 和MCM4基因在HCC 预后中的作用与有些研究不一致[38-39],可能与数据集的构成有关,需要进一步验证。
4 结论
通过在TCGA 数据集上对信号通路中41 个核心基因建模,可以对HCC 患者的预后风险进行准确分类。交叉验证表明该模型具备较高的稳健性,且在另外3 个数据集上得到准确的预测结果。将生存风险差异显著的两组进行生物信息学分析,发现HCC高风险组上调基因富集在细胞周期信号通路中,并发现了11 个hub基因的上调与HCC 风险显著相关,生物信息学分析也进一步验证模型的有效性。
