杨梅状Fe3O4@SnO2核壳材料制备及吸波性能
2020-06-05王玉江魏世丞黄玉炜徐滨士
黄 威,王玉江,魏世丞,梁 义,王 博,黄玉炜,徐滨士
陆军装甲兵学院装备再制造技术国防科技重点实验室,北京 100072
随着各种电子设备和通信技术的广泛应用,电磁波的过度辐射问题引起了人们对电磁干扰、环境污染和人类健康的极大关注[1-2].吸波材料(MAMs)可以将电磁能传化为热能,也可以通过干涉原理来耗散电磁波,因此为了防止电磁污染等相关问题的产生,人们对其进行了大量的理论和实验研究.吸波材料已被广泛应用于军事和民用领域,例如隐身防御系统、电磁屏蔽以及微波暗室等[3-4].磁铁矿Fe3O4作为一种传统的吸波材料,由于具备高饱和磁化强度,高居里温度和低成本等优良特性,被认为是最有应用前景的吸波材料之一[5].然而单一的Fe3O4具有介电损耗能力弱,易于氧化等缺陷,限制了它在微波吸收领域的发展应用[6].一般而言,良好的吸波性能与复介电常数和复磁导率的有效互补密切相关,而磁性材料和介电材料的结合可以兼顾两者磁性能和介电性能,以改良材料整体的阻抗匹配特性.因此制备Fe3O4基纳米复合材料具有广阔的前景和意义.
N型半导体材料SnO2,以其良好的气敏性、高导电性和催化性能,被广泛应用于气体传感器、阳极材料和光催化领域,更是由于其温度稳定、环境稳定的介电属性,在吸波材料领域显现其独特的优势[7-8].例如,Wang等[9]制备了一种SnO2/PPy气凝胶吸波材料,当其质量在石蜡中仅占10%时,材料的有效带宽便可达到7.28 GHz;Zhao等[10]采用酸刻蚀的方法制备了Ni@SnO2核壳结构复合吸波材料,当厚度仅为1.5 mm时,在17.3 GHz处其反射损耗可达到-50.2 dB.近年来,核壳结构的吸波材料由于具有界面极化效应、约束效应和化学均匀性,可为入射电磁波的衰减提供更多渠道,因此在提升吸波性能方面展现出巨大的潜力[11-12].受到核壳结构的启发,构建Fe3O4@SnO2能够弥补Fe3O4本身介电损耗能力弱,易于氧化的缺陷,是进一步提高吸波材料性能指标的有效途径.近年来,研究学者对Fe3O4@SnO2复合材料的研究主要集中电池及光催化领域.例如Li等[13]在Ti基底表面沉积了Fe3O4@SnO2核壳纳米薄膜,具有良好的电化学特性;Li等[14]通过水热法合成了海胆状的Fe3O4@SnO2,具有良好的光催化活性.然而,合成具有良好吸波性能的Fe3O4@SnO2核壳结构微球的报道仍然少见.
因此,本文以磁性Fe3O4微球为模板,采用Stöber法合成Fe3O4@SiO2前驱物,并采用“水热-刻蚀”的手段成功合成了一种新型的Fe3O4@SnO2复合材料,不同于之前所有的Fe3O4@SnO2核壳结构,该外壳呈杨梅状;采用X射线衍射、X射线光电子能谱、扫描电子显微镜、透射电子显微镜、振动样品磁强计研究分析了杨梅状Fe3O4@SnO2的物相结构、表面元素、微观形貌及磁性;分析比较了Fe3O4、Fe3O4@SiO2、普通Fe3O4@SnO2和杨梅状Fe3O4@SnO2的电磁损耗机制和微波吸收性能.
1 实验部分
1.1 样品制备
溶剂热法合成Fe3O4:取10 g聚乙烯吡咯烷酮(PVP)、9 g FeCl3·6H2O和36 g尿素溶解至400 mL乙二醇中,并用电动搅拌机搅拌0.5 h得到匀质的橙黄色透明溶液.随后将溶液转移至500 mL聚四氟乙烯内衬的不锈钢反应釜中,在200 ℃下保温12 h.待反应结束后,将所得产物用去离子水和乙醇各洗涤3~5次,用磁铁收集,并将最后产物在60 ℃下真空干燥10 h.
Stöber法合成Fe3O4@SiO2:取4g制备好的Fe3O4均匀分散至400 mL醇水混合物中(乙醇:350 mL,去离子水:50 mL).随后向其中依次缓慢滴加10 mL氨水和2 mL正硅酸乙酯(TEOS),并使用电动搅拌机持续搅拌8 h.最后将所得产物用去离子水和乙醇各洗涤3次,用磁铁收集,并将产物在60 ℃下真空干燥10 h.
水热法合成Fe3O4@SnO2:取2 g Fe3O4粉末均匀分散至400 mL醇水混合物中(乙醇:160 mL,去离子水:240 mL).向其中添加12 g尿素和1.8 g K2SnO3·3H2O,搅拌均匀后将溶液转移至500 mL聚四氟乙烯内衬的不锈钢反应釜中,在200 ℃下保温24 h.待反应结束后,将所得产物用去离子水洗涤6次,并用磁铁收集,最后将产物在60 ℃下真空干燥10 h,记为Fe3O4@SnO2-1.将2 g水热法合成Fe3O4@SnO2粉末按照Fe3O4@SnO2-1的合成步骤进行相同的实验操作,将所得产物记为Fe3O4@SnO2-2.
1.2 测试与表征
采用高功率转靶多晶Smartlab型X射线衍射仪(XRD)分析材料的物相(实验条件:Cu靶,步长0.02°,扫描范围10°~80°);通过Escalab 250Xi型X射线光电子能谱仪(XPS)对材料表面化学元素价态进行分析;利用Hitachi SU-8010冷场发射扫描电子显微镜(SEM)和JEM 2100F透射电子显微镜(TEM)观察材料的微观形貌;采用BHV-55型振动样品磁强计(VSM)测试材料的静磁性能;采用HP8722ES矢量网络分析仪(VNA)测量材料在2~18 GHz的复介电常数和复磁导率,测试时样品与石蜡以质量比3∶2混合,制成外径7 mm,内径3 mm,高2 mm的圆环试样;最后根据传输线理论,模拟计算不同厚度条件下材料的吸波性能.
2 结果与讨论
2.1 物相及表面元素分析
图1为Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2的X射线衍射图谱.图中位于30.2°、35.6°、43.3°、57.1°和63.1°的主衍射峰分别对应(220)、(311)、(400)、(511)和(440)晶面,与尖晶石型的Fe3O4(JCPD卡片No.99-0073)匹配良好,说明合成的Fe3O4具有很高的纯度.在Fe3O4表面包覆SiO2后,没有新的衍射峰出现,说明SiO2为非晶态[15].其余的衍射峰出现在26.6°、33.9°、51.6°和66.1°处,与金红石型的SnO2(JCPD卡片No.41-1445)相吻合,分别对应(110)、(101)、(211)和(301)晶面.对比Fe3O4@SnO2-1,Fe3O4@SnO2-2中的SnO2的衍射强度更高,表明Fe3O4@SnO2-2中SnO2的相对含量更高.
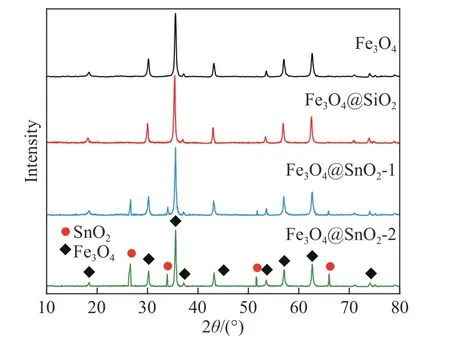
图1 不同试样的X射线衍射图谱Fig.1 XRD pattern of the studied samples
图2(a)为Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2的X射线光电子能谱总谱,Fe、O、Si和Sn等相关元素如图所示.对比Fe3O4,Fe3O4@SiO2中Fe2p的信号强度较弱,说明SiO2薄膜均匀且致密,覆盖了Fe2p信号.Fe3O4@SnO2-2中的Si2p信号(结合能为103.6 eV)几乎消失,可能是尿素在高温高压的条件下电离出了大量的OH-,通过碱性刻蚀除去了Fe3O4表面的SiO2.此外,和Fe3O4@SnO2-1相比,Fe3O4@SnO2-2中Sn3d的信号强度更强,进一步说明Fe3O4@SnO2-2中表面负载的SnO2含量更多,这和X射线衍射图谱的分析相吻合.图2(b)和(c)分别为Fe3O4@SnO2-1和Fe3O4@SnO2-2中Fe2p和Sn3d的精细谱.图2(b)中710.6和724.1 eV的峰分别对应于Fe元素中的2p3/2和2p1/2自旋轨道,两峰相差13.5 eV,其结果和文献[16]中的Fe3O4X射线光电子能谱结果吻合.结合能为715.1 eV的卫星峰来自于Sn3p3/2的干扰[17],且由于Fe3O4@SnO2-2表面的SnO2含量更高,因此反映出更强的Sn3p3/2信号干扰(如图2(c)所示).图2(d)中487.1和495.3 eV的峰分别对应于Sn元素的3d5/2和3d3/2自旋轨道,两峰相差8.2 eV,表明Fe3O4@SnO2-2中的Sn为Sn4+[18].
2.2 形貌及微观结构分析
图3为Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2的扫描电子显微镜图,右上角区域为放大后单个球体的透射电子显微镜图.所制备的Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2均为形貌规整的球形,球径在400~600 nm之间,且分散均匀,无明显团聚.图3(a)为Fe3O4的形貌及微观结构,可观察到Fe3O4球由40~80 nm的半碗状碎粒组装而成,体现了典型的奥斯特瓦尔德熟化自组装机制[19].在其表面包覆SiO2后,SiO2粒子在表面沉积成膜,膜厚大约20 nm,因此其表面比纯的Fe3O4球更为光滑(如图3(b)所示).图3(c)所示的Fe3O4@SnO2-1由Fe3O4球表面直接包覆SnO2得到,SnO2纳米粒子均匀沉积在Fe3O4球表面,形成核壳结构.另外,由于包覆过程中进一步发生了奥斯特瓦尔德熟化,因此Fe3O4@SnO2-1的具有明显的空心结构.Fe3O4@SnO2-2的表面形貌和杨梅类似(如图3(d)所示),相比于Fe3O4@SnO2-1,其形貌均匀性更好,表面SnO2负载量更多,这是因为SiO2包覆改变了Fe3O4表面性质,使后续SnO2纳米粒子在Fe3O4@SiO2模板上能够稳定沉积[20].值得注意的是,Fe3O4@SnO2-2中SnO2壳层厚度约为40 nm,壳层内部充斥大量空隙,这种结构有利于电磁波传播时多重反射及散射行为的产生.
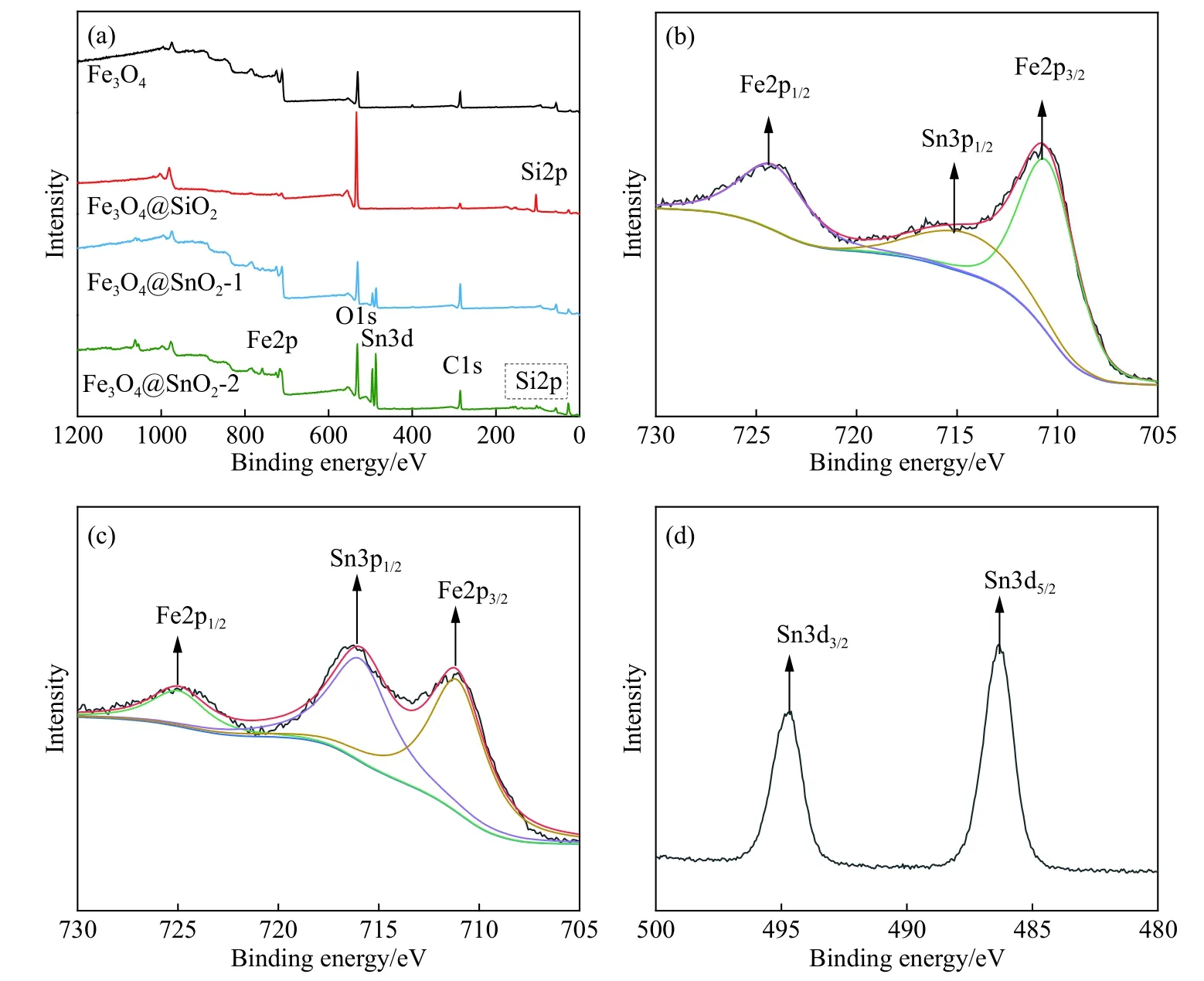
图2 不同试样的X射线光电子能谱.(a) 总谱;(b) Fe3O4@SnO2-1中的Fe2p谱;(c) Fe3O4@SnO2-2中的Fe2p谱;(d) Fe3O4@SnO2-2中的Sn3d谱Fig.2 XPS spectra of different samples: (a) XPS wide scan; (b) Fe2p spectrum of Fe3O4@SnO2-1; (c) Fe2p spectrum of Fe3O4@SnO2-2; (d) Sn3d spectrum of Fe3O4@SnO2-2

图3 试样微观结构形貌图.(a) Fe3O4;(b) Fe3O4@SiO2;(c) Fe3O4@SnO2-1;(d) Fe3O4@SnO2-2Fig.3 SEM and TEM images of samples: (a) Fe3O4; (b) Fe3O4@SiO2; (c) Fe3O4@SnO2-1; (d) Fe3O4@SnO2-2
结合X射线光电子能谱分析,反应过程中Fe3O4@SnO2-2中的SiO2薄膜由于受到碱液刻蚀,逐渐转化为硅酸盐的形式缓慢溢出.再加上SnO2纳米粒子在其周围的不断生成,因此可推知SiO2层的缓慢刻蚀可为SnO2纳米粒子的沉积提供了良好的附着环境.综上,杨梅状的Fe3O4@SnO2-2合成过程可由图4示意.
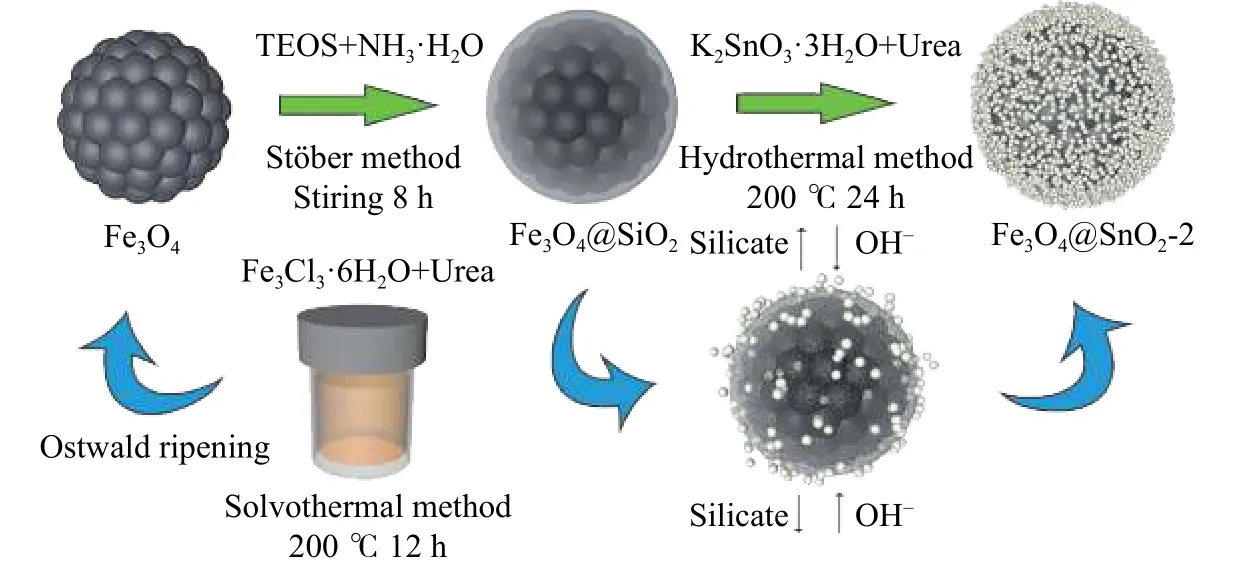
图4 Fe3O4@SnO2-2合成过程示意图Fig.4 Schematic illustration of the synthesis process of Fe3O4@SnO2-2
2.3 磁性能分析
图5为4种试样在室温下的磁滞回线.本研究制得的Fe3O4饱和磁化强度为103.9 A·m2·kg-1,矫顽力为7862.2 A·m-1,显示出一定的亚铁磁性.当Fe3O4与非磁性的壳层复合后,饱和磁化强度减弱,Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2的饱和磁化强度分别为67.5、70.2和51.2 A·m2·kg-1.虽然Fe3O4@SnO2-1和Fe3O4@SnO2-2具有相似的Fe3O4含量,但Fe3O4@SnO2-2的饱和磁化强度值要明显低于Fe3O4@SnO2-1,这是因为Fe3O4@SnO2-2的SnO2层中的空隙较多,相邻磁核距离增大,导致磁耦合性能降低.此外,由于合成Fe3O4@SnO2的水热反应过程中,奥斯特瓦尔德熟化效应继续促进了Fe3O4晶粒长大,因此Fe3O4@SnO2-1和Fe3O4@SnO2-2的矫顽力较Fe3O4和Fe3O4@SiO2有所增加[21].一般而言,初始磁导率(μi)由饱和磁化强度(MS)和矫顽力(Hc)决定,可以用来预测磁性吸波材料中的磁损耗能力,其表达式如下[22]:
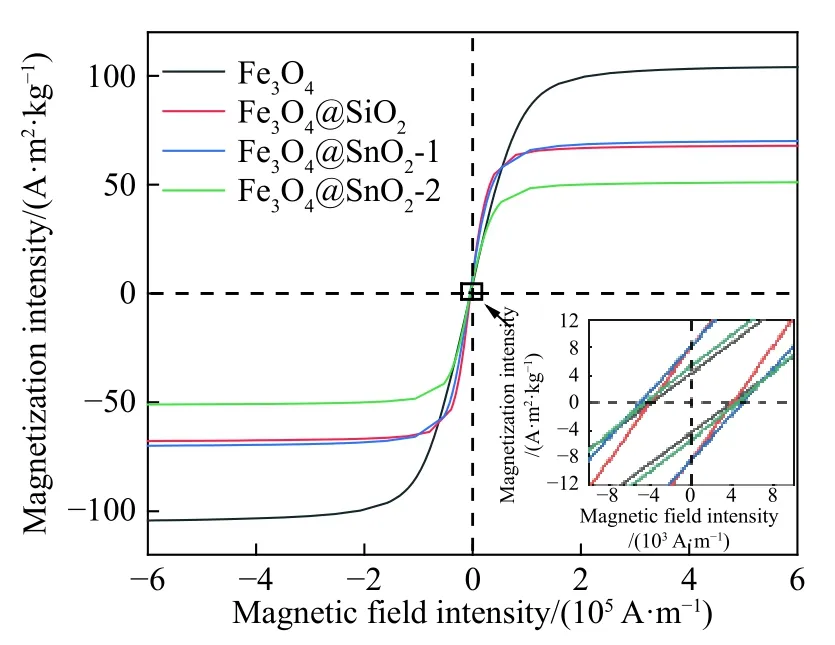
图5 试样室温下的磁滞回线Fig.5 Hysteresis loops of samples measured at room temperature
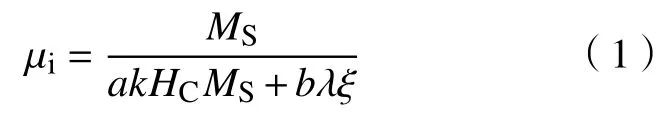
式中,a和b为由物质组成决定的两个常数,k为一个比例系数,λ为磁致伸缩系数,ξ为晶体的弹性应变参数.高的μi通常能够产生强的磁损耗,由上式可知,MS越大,Hc越小,越有利于μi的提高.基于上述磁性参数,Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2的磁损耗能力排序可能为:Fe3O4>Fe3O4@SiO2>Fe3O4@SnO2-1>Fe3O4@SnO2-2.
2.4 微波吸收性能分析
吸波材料的反射损耗(RL)可由传输线理论推导[23]:
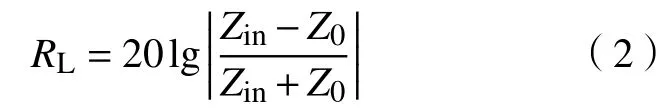
式中:Z0为自由空间阻抗,一般为1;Zin为金属背衬吸波涂层材料的输入阻抗,可由下列公式表示:
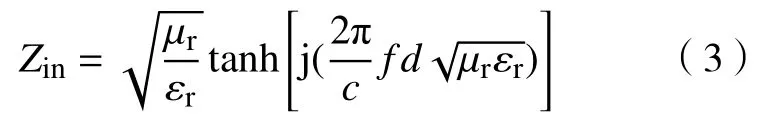
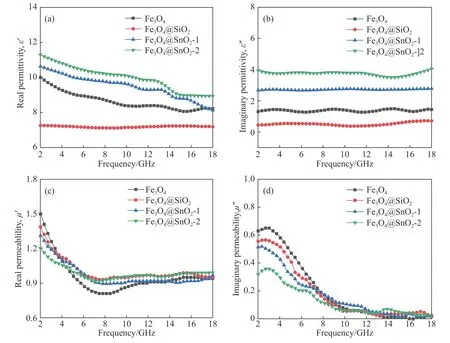
图6 试样复介电常数和复磁导率在2~18 GHz随频率变化曲线.(a) 复介电常数实部;(b) 复介电常数虚部;(c) 复磁导率实部;(d) 复磁导率虚部Fig.6 Frequency-dependent complex permittivity and complex permeability of samples: (a) real parts of complex permittivity; (b) imaginary parts of complex permittivity; (c) real parts of complex permeability; (d) imaginary parts of complex permeability
式中,εr(εr=ε'-jε")和μr(μr=μ'-jμ")分别为吸波材料的复介电常数和复磁导率.c为电磁波在自由空间的速度,f为电磁波频率,d为吸波材料的厚度.根据式(2)和(3),可知材料的吸波性能和复介电常数及复磁导率密切相关.一般而言,复介电常数和复磁导率的实部(ε'和μ')代表电磁能量的存储能力,而虚部(ε"和μ")代表电磁能量的损耗能力[24].为了分析化学成分和微观结构对材料电磁特性的影响,测量了Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2在2~18 GHz频率范围内的复介电常数和复磁导率,如图6所示.图6(a)和(b)为4种材料的复介电常数,Fe3O4的初始ε'值为9.8,随着频率增大缓慢减小,18 GHz时达到8.1,而ε"值始终在1.1~1.3之间小幅度波动.当SiO2包覆Fe3O4后,其复介电常数明显减小,这是因为SiO2绝缘薄膜将Fe3O4微球隔离开,降低了Fe3O4介电极化的程度,因此ε'值降低.同时,绝缘的SiO2薄膜降低了材料整体的电子迁移率,从而造成ε"值降低[25].与Fe3O4或者SiO2相比,SnO2具有优良的导电特性,因此不难理解Fe3O4@SnO2-1和Fe3O4@SnO2-2的介电性能要优于Fe3O4和Fe3O4@SiO2.此外,由于Fe3O4@SnO2-2表面负载有更多的SnO2,因此Fe3O4@SnO2-2的介电性能要优于Fe3O4@SnO2-1.图6(c)和(d)为样品对应的复磁导率,不同于稳定的复介电常数,Fe3O4的μ'值起始为1.5,随着频率上升剧烈下降,8 GHz时降为0.8,随后又逐渐上升,最后稳定在0.94上下.同时μ"值也在3~14 GHz从0.63下降至0.1.μ'和μ"值随频率的变化趋势表明,Fe3O4的磁损耗主要来源于自然共振损耗[26].虽然Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2也能产生类似的自然共振损耗,但由于Fe3O4的相对含量较少,因此自然共振的振幅要低于纯的Fe3O4.对比Fe3O4@SnO2-1,Fe3O4@SnO2-2中的SnO2相对含量更多,且SnO2壳层中的空隙也能充当“有效介质”,根据Maxwell-Garnet等效介质模型[27],Fe3O4@SnO2-2的复磁导率要低于Fe3O4@SnO2-1.
上述Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2对应的反射损耗可由电磁参数带入式(2)和(3)模拟计算得到.图7为它们在2~18 GHz反射损耗图,考察厚度为0~5 mm.在微波吸收领域,通常将反射损耗强度以及对应的频带作为描述吸波性能的两个重要参量.特别是反射强度在-10 dB以下的频带被视作有效吸收频带,对应电磁波的吸收率为90%~100%.此外,通常也期望吸波材料在较薄的情况下能够实现电磁波的有效吸收,这是因为若吸波涂层过厚,则在实际应用中会面临增重,结合强度低及热震性能差等一系列问题.如图7(a)所示,纯的Fe3O4在厚度超过2.5 mm时才能产生有效吸收,当厚度为3.7 mm,频率为7.6 GHz时,最小反射损耗RL(min)接近-40 dB.Fe3O4@SiO2和Fe3O4相比,虽然也能产生有效吸收,但有效吸收的区域范围和吸收强度都不如Fe3O4(如图7(b)所示).当Fe3O4与SnO2直接复合后(Fe3O4@SnO2-1),其有效吸收的区域范围大幅拓展(如图7(c)所示),通过控制吸波材料厚度1.4~5 mm,其有效吸收频带可实现在3.4~18 GHz范围内可调.Fe3O4@SnO2-1的RL(min)为-42 dB,此时厚度为2.9 mm,有效带宽为2.6 GHz(7.5~10.1 GHz).虽然Fe3O4@SnO2-2和Fe3O4@SnO2-1具有相似的有效吸收区域,但在较薄的厚度下,Fe3O4@SnO2-2在反射损耗强度上更具有优势(如图7(d)所示).当其厚度为1.4~2.8 mm时,其RL(min)均低于-20 dB,相当于至少有99%的电磁波被吸收.综合考虑吸收带宽和最小反射损耗,Fe3O4@SnO2-2的最优厚度为1.7 mm,此时RL(min)为-29 dB,其有效带宽为4.9 GHz(13.1~18 GHz).
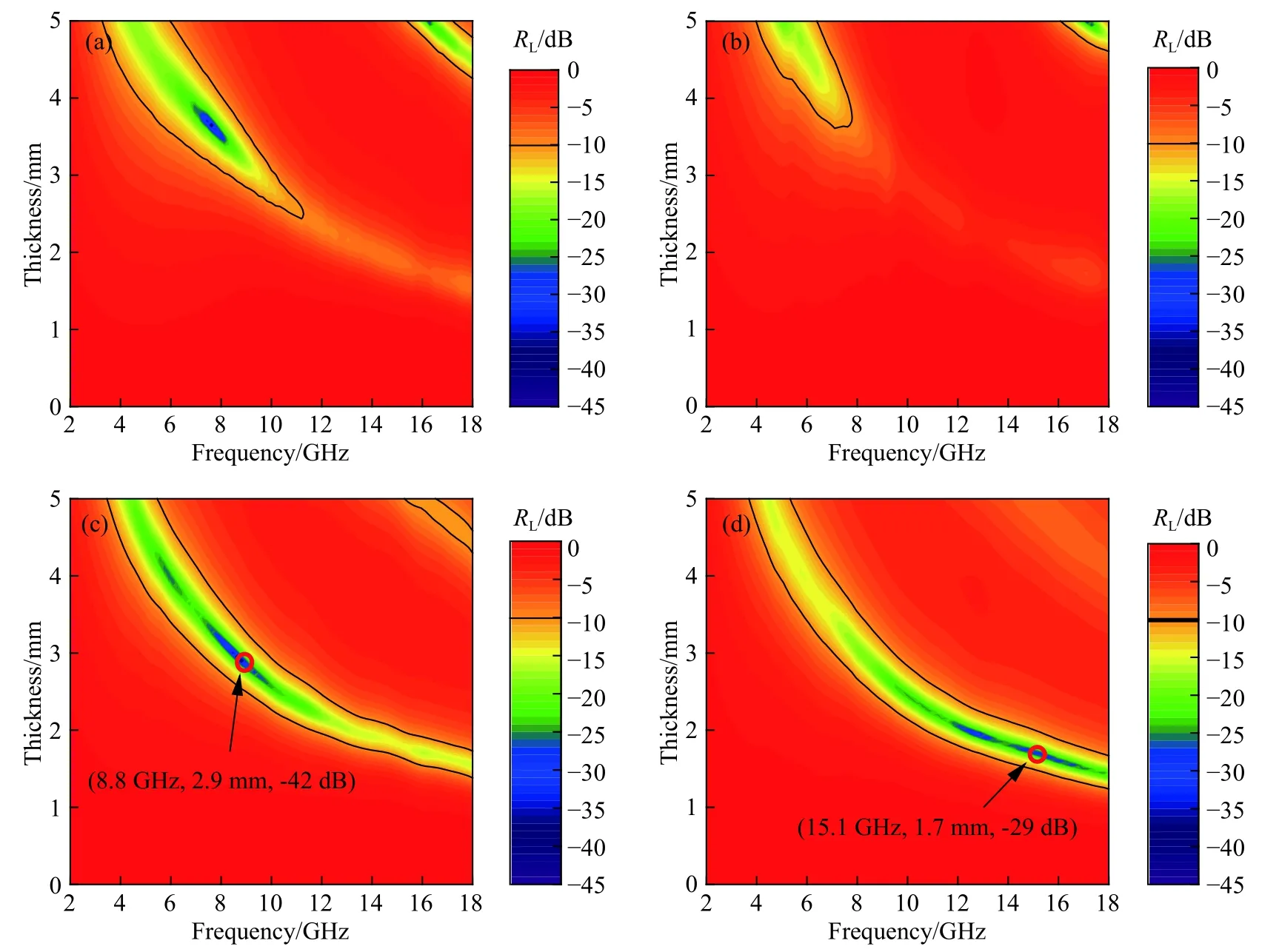
图7 试样的反射损耗图.(a) Fe3O4;(b) Fe3O4@SiO2;(c) Fe3O4@SnO2-1;(d) Fe3O4@SnO2-2Fig.7 Reflection loss maps of samples: (a) Fe3O4; (b) Fe3O4@SiO2; (c) Fe3O4@SnO2-1; (d) Fe3O4@SnO2-2
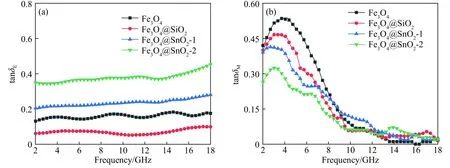
图8 试样的介电损耗正切值(a)和磁损耗正切值(b)Fig.8 Dielectric loss tangents (tanδE) (a) and magnetic loss tangents (tanδM) (b) of samples
介电损耗和磁损耗被广泛视作入射电磁波的主要损耗机制,因此采用介电损耗正切值(tanδE=ε"/ε')以及磁损耗正切值(tanδM=μ"/μ')来表征材料的介电损耗和磁损耗能力,以分析它们在电磁波损耗特性上的差异,其结果如图8所示.Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2的介电损耗和磁损耗正切值随频率变化曲线和它们的虚部曲线类似,说明虚部是影响它们损耗能力的主要因素.由于Fe3O4和Fe3O4@SiO2的ε"值很小,导致介电损耗正切值很小,因此可推断介电损耗不是它们电磁波能量损耗的主要机制.当Fe3O4与SnO2复合后,介电损耗明显增强,磁损耗呈现出不同程度的下降,表明Fe3O4@SnO2-1和Fe3O4@SnO2-2介电损耗的增强是以牺牲磁损耗为代价的.和Fe3O4@SnO2-1相比,Fe3O4@SnO2-2的介电损耗能力更强,这主要归因于Fe3O4@SnO2-2表面的SnO2负载量的提高增强了材料的导电损耗能力;其次,杨梅状Fe3O4@SnO2-2的SnO2层内空隙能提供更多的散射位点,使入射电磁波产生强烈的多重散射行为,从而增强材料对入射电磁波的能量消耗.另外,与介电损耗相对应,Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2磁损耗能力大小排序和前文磁性能分析中所推测的排序相同,表明这些材料的磁损耗能力只取决于磁响应能力和磁相互作用,而不是其他因素.
除了介电损耗和磁损耗,吸波性能还取决于电磁波的相消干涉,可用四分之一波长模型解释[28],即:
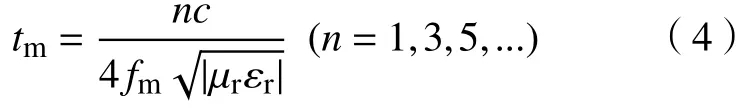
式中,tm为出现损耗峰时吸波材料的厚度,fm为损耗峰的峰值频率.当电磁波垂直入射到金属背衬的吸波涂层材料时,一部分电磁波会在空气-涂层界面被反射回来;而进入吸波涂层的电磁波会在涂层内部继续传播和损耗,接触金属背衬时会被完全反射,与之前反射的电磁波相互干涉.要形成损耗峰,即两束电磁波发生相消干涉,吸波材料的厚度需满足电磁波在介质中波长λ的四分之一及其奇数倍,此时两束电磁波的相位差刚好为180°.此外,文献[29]还指出,损耗峰的强度由两束电磁波的能量差决定,即当两束电磁波能量相接近时才能形成较强的损耗峰,此时吸波材料的归一化特征阻抗(Z=Zin/Z0)应接近1,否则即使满足相消干涉的条件也只能形成较弱的损耗峰.图9为Fe3O4、Fe3O4@SiO2、Fe3O4@SnO2-1和Fe3O4@SnO2-2归一化特征阻抗Z分布图,并绘制了相应的λ/4厚度-频率曲线.显然,这4种材料具有相似的λ/4厚度-频率曲线,不同厚度下损耗峰的峰值频率和该曲线的坐标位点相吻合,其损耗峰的强度完全由Z值决定.Fe3O4的Z值接近1的区域很小,表明其输入阻抗与自由空间阻抗的匹配性较差,而当绝缘的SiO2包覆后,使Fe3O4原本的较弱介电损耗能力进一步降低,因此造成更严重的阻抗失配,导致吸波性能进一步下降,其结果和图7(a)和(b)所示的吸波性能分析相吻合.当Fe3O4与SnO2复合后,介电损耗能力增强,磁损耗能力减弱,两者达到了一个相对平衡的状态,因此阻抗匹配能力得到提升.另外,Fe3O4@SnO2-2中Z值接近1的区域更集中于低厚度区,说明相比于Fe3O4@SnO2-1,Fe3O4@SnO2-2更容易在较薄的厚度下实现阻抗匹配.基于以上分析,杨梅状的Fe3O4@SnO2-2不仅具有较强的介电损耗能力,且有利于提升阻抗匹配性能,因此表现出更好的电磁波吸收能力.
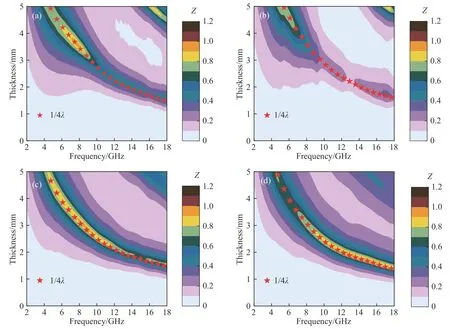
图9 研究试样的阻抗匹配图.(a) Fe3O4;(b) Fe3O4@SiO2;(c) Fe3O4@SnO2-1;(d) Fe3O4@SnO2-2Fig.9 Impedance matching maps of studied samples: (a) Fe3O4; (b) Fe3O4@SiO2; (c) Fe3O4@SnO2-1; (d) Fe3O4@SnO2-2
3 结论
(1)以空心Fe3O4微球为模板,采用Stöber法合成了Fe3O4@SiO2前驱物,并采用水热法进一步合成了杨梅状的Fe3O4@SnO2.X射线衍射和X射线光电子能谱结果显示杨梅状的Fe3O4@SnO2晶型良好,合成纯度较高.
(2)杨梅状的Fe3O4@SnO2呈球状,球径约为500 nm,且分散均匀无明显团聚.其SnO2层由纳米SnO2颗粒松散堆叠而成,具有大量的空隙结构,层厚约为40 nm.
(3)相 比 于Fe3O4@SnO2-1,Fe3O4@SnO2-2具有更好吸波性能,归因于介电损耗的增强和良好阻抗匹配.杨梅状的Fe3O4@SnO2最优厚度为1.7 mm,此时RL(min)为-29 dB,有效带宽为4.9 GHz(13.1~18 GHz),是一种具有发展潜力的吸波材料.
