SPE-UPLC-MS/MS直接测定食品中草甘膦及其代谢物氨甲基膦酸
2020-06-03孙文闪章虎张安平诸骏杰余鹏飞沈雄雅
孙文闪,章虎,张安平,诸骏杰,余鹏飞,沈雄雅
1. 绿城农科检测技术有限公司(杭州 310051);2. 浙江工业大学环境学院(杭州 310014)
草甘膦(Glyphosate,Gly)作为无选择类除草剂,以其高效、低毒、廉价等特性被广泛运用于全球各个农业和非农业领域[1-2]。草甘膦在环境和生物体中富集并通过食物链进入人体,进而对人体健康造成危害。随着草甘膦在农业生产中的广泛使用,其及其主要代谢产物氨甲基膦酸(Aminom ethyl phosphonic acid,AMPA)残留问题也越来越受关注。GB 2763—2016《食品安全国家标准食品中农药最大残留限量》对草甘膦的最大残留量有如下规定:谷物0.1~0.5 mg/kg,水果0.1~0.5 mg/kg,茶叶1 mg/kg。草甘膦及其代谢产物氨甲基膦酸都具有易溶于水,难溶于一般有机溶剂,难挥发,缺少发色和荧光基团等特性,因此运用常规方法进行检测比较困难[3]。目前,草甘膦及其代谢物氨甲基膦酸检测方法有气相色谱(GC)法[4]、离子色谱(IC)法[5-8]、液相色谱(LC)法[9-10]、气相色谱质谱(GC-MS)法[11]、液相色谱质谱(LC-MSMS)法[12-17]、电感耦合等离子体质谱仪(ICP-MS)[18]等,但是这些方法大部分需要衍生反应,时间长甚至要过夜,操作程序繁琐,稳定性差,灵敏度低,有机试剂污染严重,难以直接运用在食品样品的检测中。因此建立一种简单、便捷、有效的草甘膦及其代谢物残留的检测方法具有重要的意义。试验建立了SPEUPLC-MS/MS直接测定食品中草甘膦及其代谢物氨甲基膦酸的方法,该方法简便、快捷、有效,前处理步骤少,有机溶剂消耗量小,无需使用酸碱试剂调节pH,样品无需衍生,结果稳定性高,可满足各类食品的检测需求。
1 试验部分
1.1 主要仪器与试剂
LC30A液相色谱(日本岛津公司);ABSCIEX QTRAP 5500质谱系统(美国AB SCIEX公司);KQ5200E超声波清洗器(昆山舒美);ST16R高速离心机(美国赛默飞世尔公司);EUFO-945616涡旋震荡器(TALBOYS);Milli-Q型超纯水仪(美国密理博公司);BSA224S电子天平(德国赛多利斯公司);Oasis HLB固相萃取小柱(5 mL/200 mg,Waters公司);0.22 μm水溶性滤膜(上海安谱实验科技股份有限公司);1 mL医用注射器(岛津技迩(上海)商贸有限公司);50 mL离心管(美国赛默飞世尔公司)。
乙腈、二氯甲烷(色谱纯,德国CNW科技公司);乙酸铵(质谱纯,美国赛默飞世尔公司);草甘膦以及氨甲基膦酸标准品(纯度>98%,德国Dr Ehrenstofer公司)。样品市售。
1.2 样品前处理
提取:称取2 g(精确到 0.001 g)样品于 50 mL聚丙烯离心管中,加入10.0 mL超纯水,涡旋混合3 min,超声提取20 min,再加入5 mL二氯甲烷,涡旋混合2 min,以 8 000 r/min离心 5 min,上清液待净化。
净化:取2 mL上清液,过HLB-SPE小柱净化,HLB-SPE小柱先经过5 mL的纯水和5 mL甲醇活化,前1 mL上清液过柱,弃去不要,后1 mL过柱后收集,过0.22 μm水溶性滤膜,待上机。
1.3 仪器条件
液相色谱条件:色谱柱Waters BEH Amide氨基柱(2.1 mm×50 mm,1.7 μm);柱温35 ℃;进样量5 μL;流速0.25 mL/min;流动相,A-乙腈,B-5 mmol/L乙酸铵溶液,氨水调节至pH 11,洗脱程序见表1。
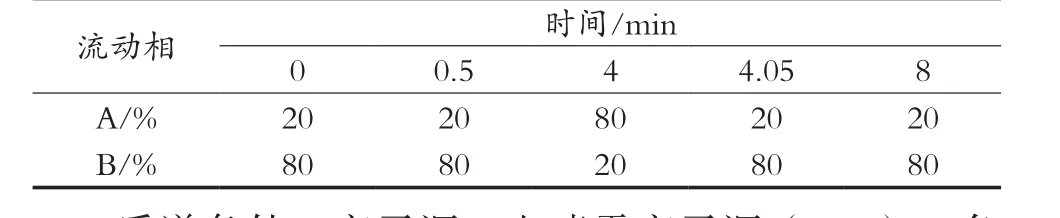
表1 洗脱程序
质谱条件:离子源,电喷雾离子源(ESI),负离子模式;离子源温度550 ℃;喷雾电压-4 500 V;气帘气流量40.0 psi;喷雾气流量60.0 psi;辅助加热气流量60.0 psi;扫描方式,多反应监测(MRM);监测离子对见表2。
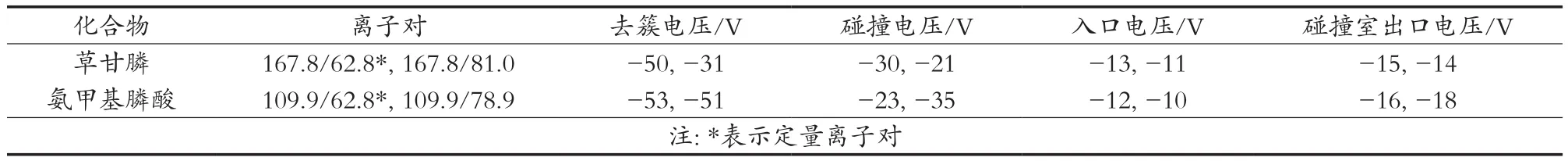
表2 草甘膦以及氨甲基膦酸的质谱参数
1.4 标准溶液配制
分别称取草甘膦以及氨甲基膦酸标准品,用纯水溶解配制成200 μg/mL标准储备液,在4 ℃下保存待用。
标准曲线的配制:采用空白样品(绿茶、大米、小麦粉、草莓、白萝卜),经1.2提取净化,配制成5个不同浓度基质标样(5.00~100 μg/L和10.0~200 μg/L),现用现配。
2 结果与讨论
2.1 质谱条件的确立
草甘膦及其代谢产物氨甲基膦酸具有较强的极性,均具有易溶于水的特性,在水中能够解离生成少量的氢离子,属于较弱的有机酸,因此适合采用负离子源模式检测,选择ESI负离子模式对草甘膦和氨甲基膦酸进行母离子扫描(一级扫描),得到草甘膦和氨甲基膦酸的母离子,分别为167.8和109.9;然后再进行子离子扫描(二级扫描),得到的草甘膦的子离子(62.8和81.0),氨甲基膦酸的子离子为62.8和78.9;再对这些离子对进行碰撞电压、去簇电压、入口电压和碰撞室出口电压的优化,得到最佳的响应条件,见表2。最后对离子源温度、喷雾电压、气帘气流量、喷雾气流量、辅助加热气流量进行优化,得到最佳的离子源参数:离子源温度550 ℃;喷雾电压-4 500 V;气帘气流量40.0 psi;喷雾气流量60.0 psi;辅助加热气流量60.0 psi。
2.2 液相条件的选择
质谱检测虽然具有高的灵敏度和选择性,但是对草甘膦和氨甲基膦酸的强极性和易溶于水的物质,需要选择合适的色谱柱、流动相以及洗脱条件,使其具有尖锐对称的峰型和一定的分离度,才能发挥出质谱的优势。常规的C18和T3等色谱柱对草甘膦和氨甲基膦酸没有保留,于是选择HILIC模式色谱柱对其进行分离,氨基柱是HILIC模式中的一种,在糖类等碳水化合物分析中广泛应用[19-20],于是尝试用它来分析,氨基柱常用的流动相是乙腈和水,由于草甘膦和氨甲基膦酸采用的是负离子模式,在流动相中可以添加适当浓度的氨水和乙酸铵来提高离子化效率而提高响应,同时改善色谱峰的峰型,缓冲体系进一步增加系统的稳定性,试验发现乙酸铵的浓度为5 mmol/L,峰型尖锐对称,又不会对检测器造成太多的污染,故选择5 mmol/L乙酸铵,当乙酸铵的浓度为5 mmol/L时,再添加不同浓度的氨水调节乙酸铵溶液的pH,草甘膦和氨甲基膦酸的响应与乙酸铵溶液pH的关系见图1。结果显示,pH越高,响应越好,这是因为在碱性条件下,草甘膦和氨甲基膦酸更容易形成[M-H]-准分子离子,但是考虑Waters BEH Amide氨基柱pH上限,最终确定乙酸铵溶液的pH为11。同时梯度洗脱有助于降低基质对目标化合物检测干扰,同时可有效减少杂质对色谱柱的污染,获得更好的分离效果和质谱响应,故选择梯度洗脱。试验得到的最佳液相条件为:色谱柱Waters BEH Amide氨基柱(2.1 mm×50 mm,1.7 μm),流速0.25 mL/min,流动相pH 11的5 mmol/L乙酸铵溶液和乙腈梯度洗脱,洗脱程序见表1。在该条件下进样分析,草甘膦和氨甲基膦酸基质标准溶液的色谱图见图2,空白样品的色谱图见图3,空白样品添加回收的色谱图见图4。从图2~图4可以看出,优化后的液相条件可以得到尖锐对称的峰型,无杂质干扰,可以进行准确的定量计算。
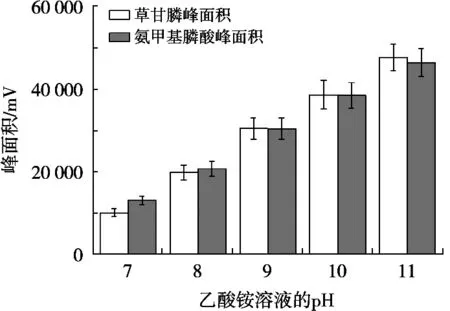
图1 草甘膦和氨甲基膦酸的峰面积与乙酸铵溶液的pH关系图
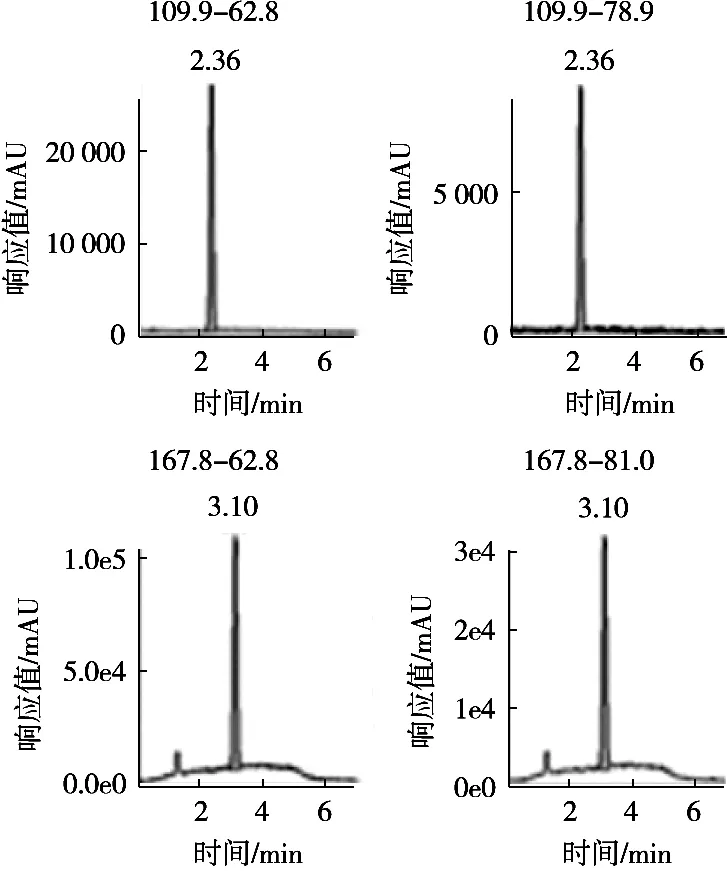
图2 草甘膦以及氨甲基膦酸基质标准溶液LC-MS-MS色谱图
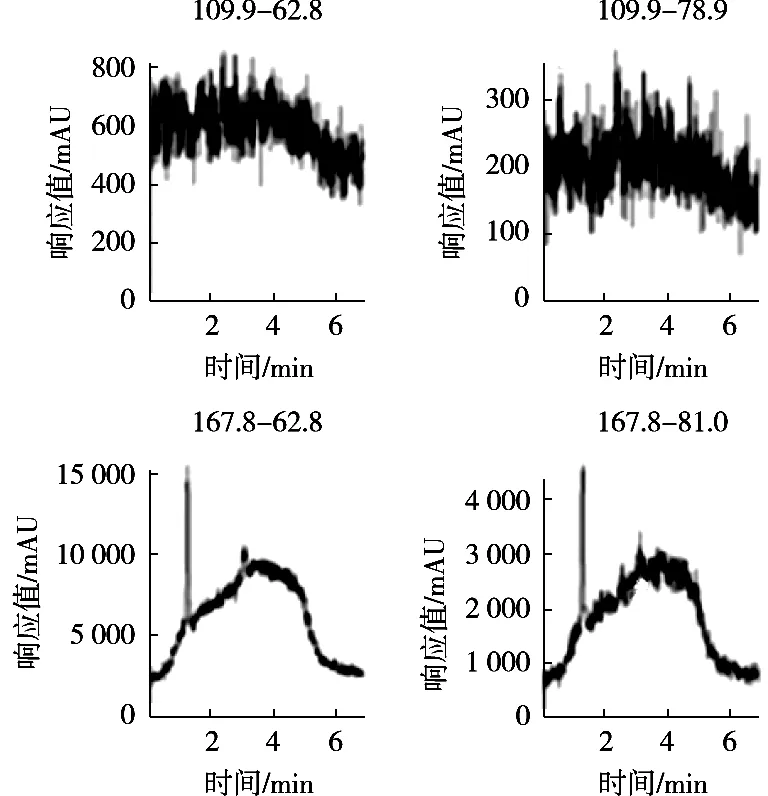
图3 空白样品的LC-MS-MS色谱图
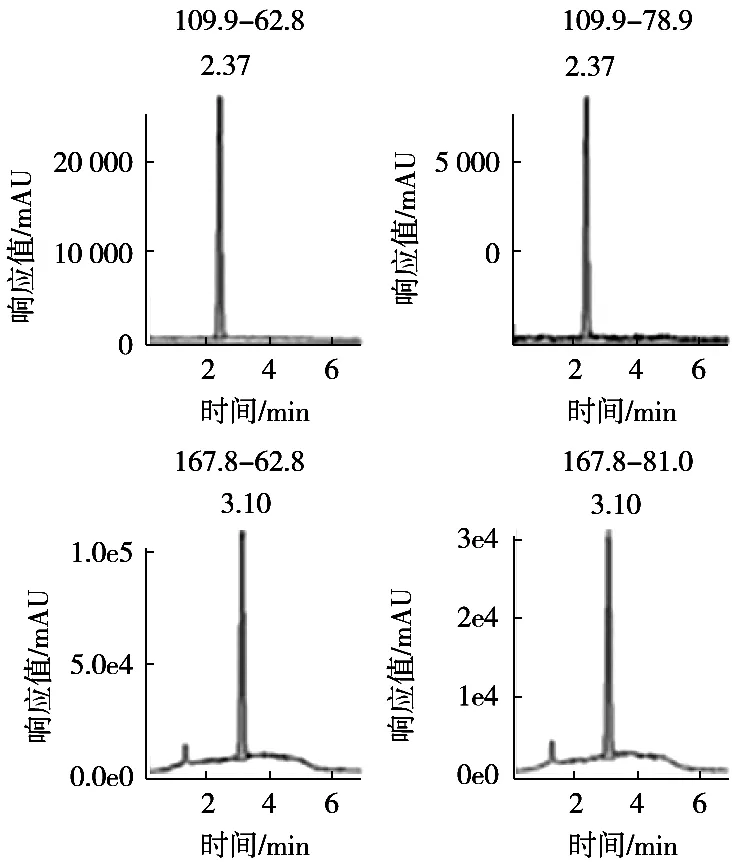
图4 空白样品添加回收的LC-MS-MS色谱图
2.3 样品的净化
样品用水提取后,首先通过二氯甲烷萃取除去脂溶性色素以及其他一些非极性杂质,然后再用固相萃取小柱进行净化。Oasis HLB小柱的填料为亲水亲脂的反相吸附剂,可以有效去除蛋白质、脂肪、色素等物质,在抗生素[21]和激素[22]等检测领域得到广泛的应用,因此选择Oasis HLB小柱作为净化柱。分别将10.0和50.0 μg/L草甘膦和氨甲基膦酸标准溶液用HLB净化,其回收率在93.8%~102%之间,说明HLB小柱对草甘膦和氨甲基膦酸没有吸附,因此可以采取过滤式净化模式,不需要清洗和洗脱步骤,操作简单。对茶叶提取液过柱前后成分进行测定,发现过柱后茶多酚减少了95.2%,咖啡因减少了93.6%,氨基酸减少了62.6%,说明HLB小柱除了可以除去蛋白质、脂肪、色素等大分子物质,对一些小分子物质也有较好的除去效果,因此采取Oasis HLB固相萃取柱进行净化。
2.4 方法的检出限、定量限、线性范围
方法检出限(LOD)的确定通过空白样品进行加标并不断降低加标浓度,进样分析信噪比为3,通过试验和分析得到草甘膦和氨甲基膦酸的检出限分别为10.0和20.0 μg/kg。定量限(LOQ)的确定通过空白样品进行加标并不断降低加标浓度,进样分析信噪比为10,并且回收率在70.0%以上,通过试验和分析得到定量限分别为25.0和50.0 μg/kg。在1.3仪器条件下,草甘膦以及氨甲基膦酸分别在5.00~100 μg/L和10.0~200 μg/L质量浓度范围内有良好的线性关系,绿茶、大米、小麦粉、草莓、白萝卜五种基质标准曲线线性方程和相关系数见表3。
2.5 方法的准确度与精密度
方法的准确度通过空白样品添加回收试验的回收率来考察,添加浓度为定量限、2倍定量限和10倍定量限;精密度通过同一浓度平行添加6次回收率的相对标准偏差(RSD)来获得,不同样品基质回收率和相对标准偏差(RSD)的结果见表4。可以看出:该方法中草甘膦和氨甲基膦酸在各种基质中的回收率在72.0%~90.8%之间,精密度RSD值小于10.0%,符合GB/T 27404—2008《实验室质量控制规范基本信息 食品理化检测》中的回收率和精密度的要求。
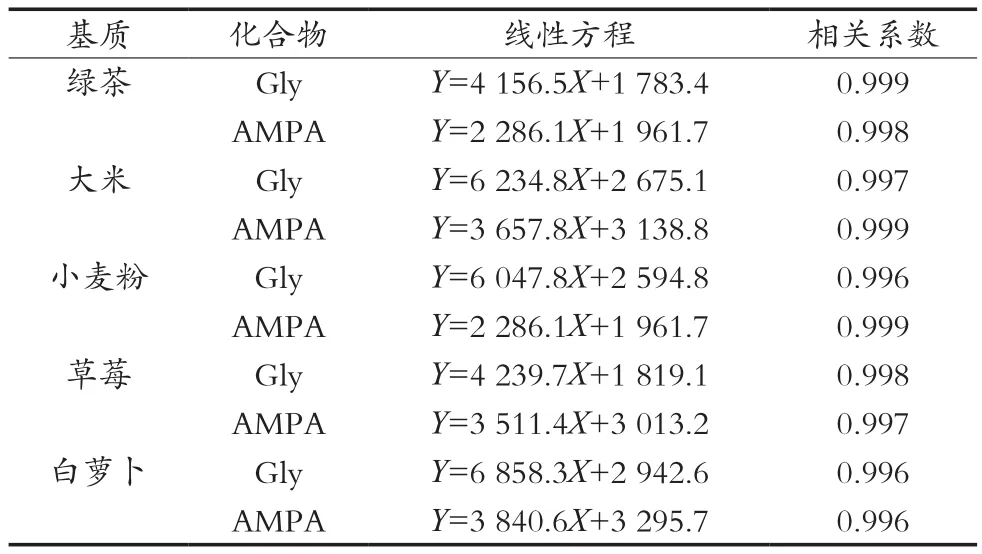
表3 五种基质标准曲线线性方程和相关系数
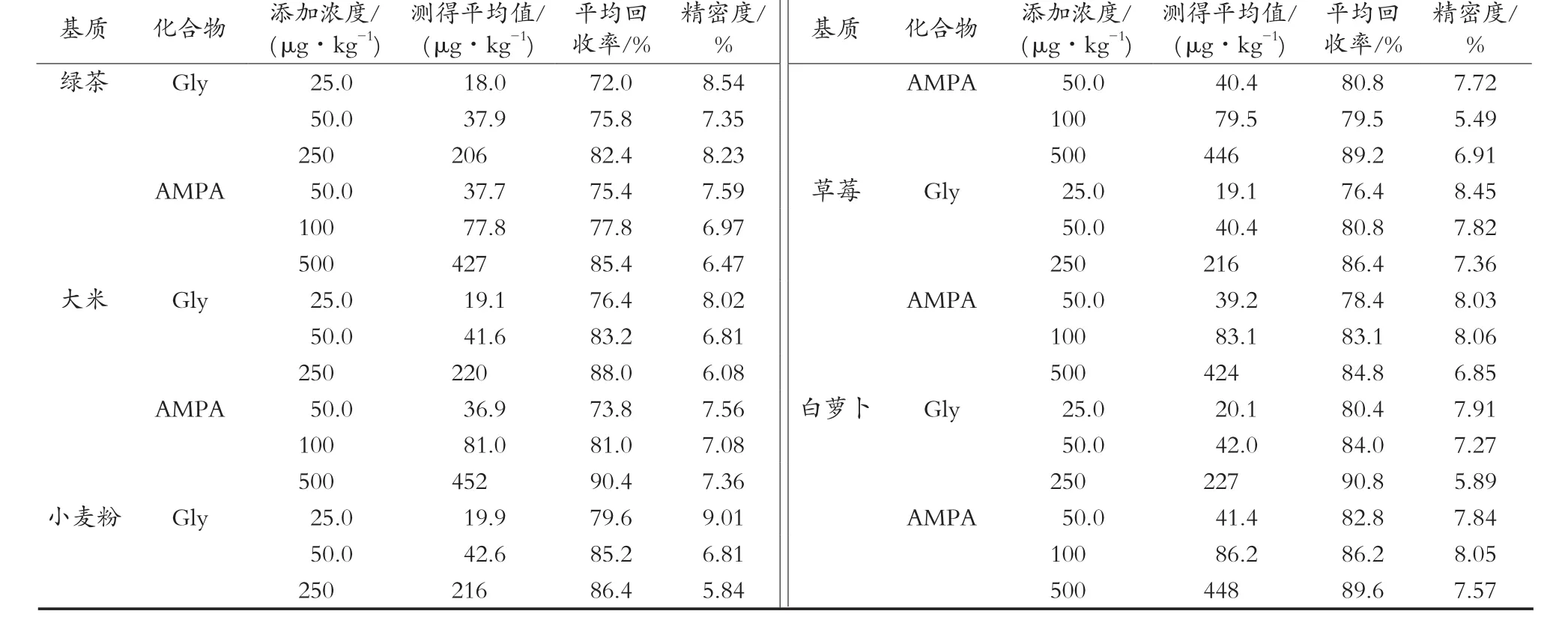
表4 回收率和精密度试验结果
3 实际样品测定
运用此方法对市场上55份食品进行检测,其中茶叶样品22份、大米9份、小麦粉5份、水果12份、蔬菜7份结果显示,茶叶样品检出5份,含量在0.062~1.31 mg/kg之间;水果样品中草莓检出1份,含量为1.45 mg/kg;大米检出1份,含量为0.064 mg/kg。该方法可以满足GB 2763—2016《食品安全国家标准食品中农药最大残留限量》对草甘膦的最大残留量的规定,为食品中相关标准的修订和制定提供借鉴意义。
4 结论
试验通过采用水提取,HLB小柱净化,氨基色柱进行分离,UPLC-MS/MS测定食品中草甘膦及其代谢物氨甲基膦酸。该方法简便、快捷、有效,前处理步骤少,无需使用酸碱试剂调节pH,样品无需衍生,结果准确性好,可满足各类食品的检测需求,具有一定的推广价值。
