基于 XGBoost 的基因静态数据调控网络推断方法
2020-06-03车丹丹姜青山
车丹丹 郭 顺 姜青山
1(中国科学院深圳先进技术研究院 深圳 518055)
2(中国科学院大学深圳先进技术学院 深圳 518055)
1 引 言
解析基因调控网络(Gene Regulatory Networks,GRNs)的结构对生物信息学至关重要,因为它为生物有机体的发展机理及功能等提供了一个新的研究视角。随着微阵列技术的发展,全基因组范围上的基因表达都可被观测到,这为从基因表达数据上推导调控网络拓扑结构提供了契机。基因调控建模是根据基因表达数据所蕴含的信息而建立的反映基因与基因之间调控关系的网络,而重构基因调控网络是后基因组时代非常重要的研究领域[1]。基因调控网络对所有生物种类和系统的作用是显而易见的,因为其在维持生物有机体的功能方面发挥着重要作用[2]。因此,重构基因调控网络有着广泛的应用前景,它为药物设计和医疗相关领域等提供了重要信息。
基因是遗传信息的基本载体。虽然一个有机体中的所有基因都是相同的,但它们可以根据基因间的相互作用及网络在不同组织中准确表达并执行特定的功能[3]。在研究和认识基因之间及相应网络的相互作用工作中,重构的基因调控网络可作为一种工作模型,为研究者在实验设计上提供辅助和形成新的假说。例如,Hecker 等[4]将重构基因调控网络应用于卵巢癌,产生了一系列可检验的假说,并发现了一个潜在的药物靶点。Camacho 等[5]提出机器学习和网络生物学相结合的交叉学科有望在疾病生物学、药物发现、微生物研究和合成生物学等领域取得重大突破。
Boosting 方法是一种强大且常用的统计学习方法,主要贡献在于可以将弱学习算法提升为强学习算法,具有许多传统方法所没有的优点,故其在基因表达数据上的应用十分广泛。在分类学习中,Boosting 方法通过反复修改训练数据的权值分布,构建一系列基本分类器,并将这些分类器进行线性组合,构成一个强分类器。对于回归问题,Boosting 方法通过多次对训练样本做重抽样建模,学习多个回归器,并将这些回归器进行组合,可较大幅度提高回归模型的性能[6]。
本文针对静态基因表达数据,致力于研究基因调控网络重构方法,目标在于构建具有更好可靠性及准确率的基因调控网络。具体地,针对真实的静态基因表达数据,建立特征选择集成框架,选择 Boosting 模型计算基因调控关系的初始权重,并在初始排序基础上增加归一化和统计方法以提高模型准确率。
2 基因调控网络推断方法研究现状
调控网络已广泛应用于各种生物系统的结构建模,且已开发出一系列方法来构建可靠的生物网络[7]。Lee 和 Tzou[8]通过分析从基因表达数据上推导调控网络的不同计算方法,表明现有方法均具有不同程度的准确性和复杂性,虽然实用性已有所提高,但准确率仍待提高。由于是从基因表达数据来推导调控网络拓扑结构,因此这些方法被称为基因调控网络的逆向工程(Reverse-Engineering),也可称为重构基因调控网络(Reconstructing GRNs)。
按照时间顺序,国内外学者从基因表达数据上推导调控网络拓扑结构的问题研究工作主要有 4 个阶段(见图 1):基于统计分析的方法[9]、基于信息论的方法[10-12]、基于概率图模型的方法[13-17]和基于机器学习的方法[18-26]。

图 1 现有研究方法发展历程Fig. 1 Development of existing research methods
2.1 基于统计分析的方法
1989 年,一种基于统计分析的重构基因调控网络方法被提出[9],该方法使用相关系数来定义基因之间的相似性度量并以此推导基因网络。这种方法的主要缺点是相关系数很难识别基因之间更加复杂的依赖关系。
2.2 基于信息论的方法
为解决基于统计分析方法的局限性,研究者提出了一些基于信息论的方法——使用基因之间的互信息(Mutual Information)作为度量并以此推导调控网络[10-12]。由于在相关网络中,一般存在间接调控关系,故需采用一些方法来消除间接调控关系的影响。例如,CMI2NI[10]通过计算包含和排除两个基因之间边缘的假设分布之间的 Kullback-Leibler 差异,来量化给定两个基因之间的相互信息;BC3NET(Bagging C3NET)[11]使用基于对互信息值连同最大步长(Maximization Step)的估计来进一步提高所重构基因调控网络的识别率;ANOVerence[12]引入元信息(Meta-Information)来推导调控网络并使用基于相关系数的评分来估计基因之间的依赖关系。以上仅基于信息论的方法均存在一个缺点——识别率有限,对于大量冗余和不相关特征无法做有效识别。
2.3 基于概率图模型的方法
自 2000 年起,许多基于概率图模型的方法(如贝叶斯网络方法)被广泛应用于重构基因调控网络。Friedman 等[13]首次提出了基于贝叶斯网络来表示统计依赖关系的框架——利用贝叶斯网络学习工具从微阵列数据中恢复基因相互作用;Liu 等[14]提出了局部贝叶斯网络方法,利用网络分解策略和伪正边缘消除方法从基因表达数据中推断 GRNs。DBNCS(Dynamic Bayesian Network Comprehensive Score)算法将综合评分与动态贝叶斯模型相结合,首次构造出具有多重时延的 GRNs[15];Xing 等[16]提出了一种基于互信息和断点检测的候选自动选择算法(CAS)来限制搜索空间,以加速贝叶斯网络的学习过程,但效率仍低于同期部分其他方法。De Campos 等[17]利用结构约束的贝叶斯网络学习算法来对基因表达数据构建 GRNs。
然而,静态贝叶斯网络方法只能构建无环网络,并需将数据进行离散化处理从而造成信息丢失,而动态贝叶斯网络方法则通常局限于在时间序列数据上的应用。另外,无论是从理论上还是从计算效率上来看,贝叶斯网络的结构学习都是一项很大的挑战,尤其是当该类方法应用于维度特别高的基因表达数据的时候。
2.4 基于机器学习的方法
自 2009 年起,出现了许多对重构基因调控网络方法比较评估方面的研究。其中,DREAM(Dialogue for Reverse Engineering Assessments and Methods)挑战[18]为相关领域的研究者们提供了基准数据集来验证评估他们的工作。其中,基于特征选择框架的集成方法在 DREAM 挑战基准数据集上有较为突出的识别率。例如,GENIE3(GEne Network Inference with Ensemble of Trees)[19]在学习过程中使用随机森林(Random Forests)的特征选择方法,但该方法在理论上不具有较好的可理解性。为此,TIGRESS[20]在学习过程中使用最小角回归方法(Least Angle Regression)的特征选择方法并结合稳定性选择(Stability Selection)来解决重构基因调控网络问题。NIMEFI(Network Inference Using Multiple Ensemble Feature Importance Algorithms)[21]考虑了不同基于特征选择框架的集成方法的互补性,将 GENIE3、E-SVR(Ensemble Support Vector Regression)以及 E-EL(Ensemble Elasitc Net)等方法置于统一的框架下进行学习,并以此解决重构基因调控网络问题。然而,NIMEFI 最大的问题在于其所需的参数远大于其他方法,这给模型的参数选择带来很大的挑战,并且极大地增加了该模型的不确定性。Guo 等[22]利用基于偏最小二乘的线性方法对 GRNs 进行建模,但在真实实验的数据集上该线性模型仍存在明显局限性,模型准确率低;Chi 和 Liu[23]利用基于模糊逻辑和神经网络的认知模糊影响图(FCMs)对 GRNs 进行建模,但预测结果准确率不高;Deng 等[24]利用微分方程进行网络推断,并引入了一个具有自适应数值微分的线性微分方程模型,该模型可扩展到非常大的调节网络;Petralia 等[25]提出了一个灵活统一的集成框架,允许将来自异类数据的信息共同考虑用于 GRNs 推断;Zheng 等[26]提出了一种结合互信息的集成框架,首先对候选调控基因进行预加权,然后利用 MARS(Multivariate Adaptive Regression Splines)检测非线性调控链,但该方法存在模型过拟合问题,无法准确提取基因调控关系。
由于基因表达数据通常含有大量冗余和不相关特征,而现有基于机器学习的模型或结构复杂参数过多导致过拟合严重,模型不稳定;或使用线性模型等简单模型进行拟合,效果差、准确率低。本文提出一种基于 Boosting 集成模型的方法[6](XGBoost),应用随机化和正则化来解决模型过拟合问题,同时针对不同子问题建模所得权重不一致问题,对初始权重增加归一化和统计学方法处理。
3 基于 XGBoost 的基因静态数据调控网络推断方法

(2)学习过程:为了识别所有潜在的调控关系,学习过程将每个子集定义为统计上的特征选择问题。对于小样本的数据,直接通过特征选择方法来求解效果并不理想,本文选择 XGBoost 模型,通过改变训练样本的权重,同时学习多个基模型,并将多个基模型线性组合以提高性能。
(3)融合过程:该过程将学习过程得到的每个子集的调控关系进行融合,最终将所有的调控关系根据权重大小进行排序。对于不同子问题分别建模得到的权重,存在量纲不一致问题,本文在初始排序基础上增加归一化和统计方法以提高模型准确率。
3.1 分解过程
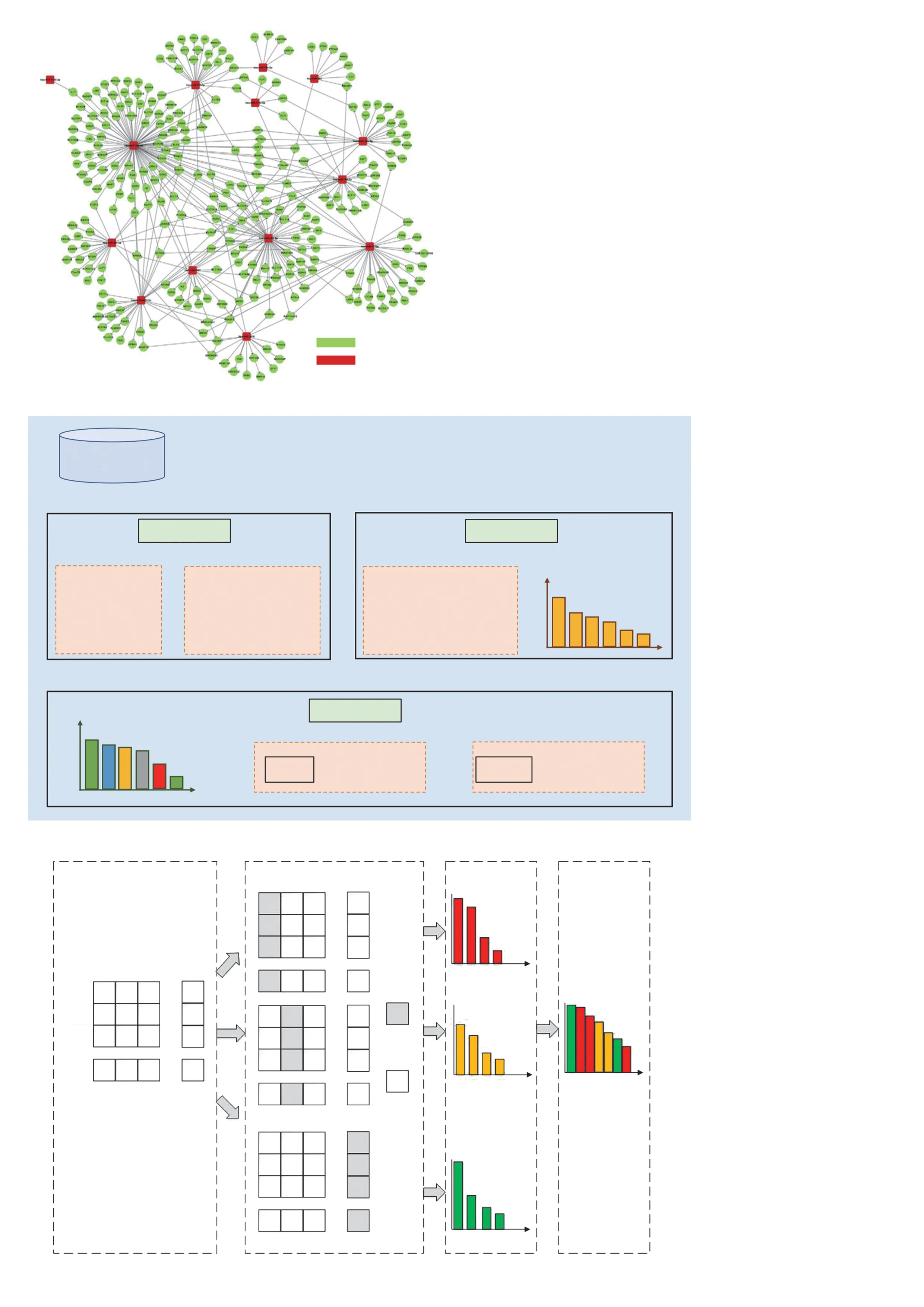
图 2 调控网络推断方法流程图Fig. 2 Flow chart of regulation network inference method
图 3 基因表达数据处理流程图Fig. 3 Gene expression data processing flow chart

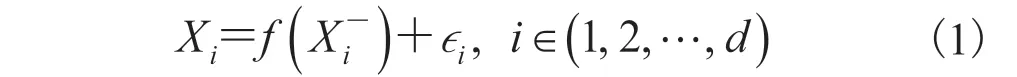
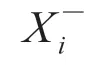
3.2 学习过程
本文选择 Boosting 模型评估基因调控关系的重要性,多次对训练样本做重抽样并建模,学习多个回归器,并将这些回归器进行组合,提高回归性能。由于传统的 Boosting 集成学习方法需要学习多个弱学习器,训练时间相对较长,而 XGBoost 模型[6]使用二阶导数的信息来帮助迭代训练,损失函数值将能更快地下降,提高训练速度,获得高性能模型。XGBoost 的目标函数可以表述为:
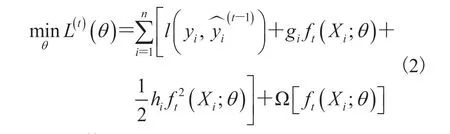
其中,T 为树结构中的树的叶子节点个数; 和 λ 为控制收缩的参数;w 为叶子权重。正则化项可以对最终学习得到的权重进行平滑,即 XGBoost 模型通过 LASSO(L1)和 Ridge(L2)正则化来惩罚更复杂的模型,从而避免过拟合问题。
本文应用特征变量 Gi的个数 Ni来分割所有树结构中的目标变量,作为 Gi的重要性。分割标准和其他细节可参考文献[6]。针对静态表达式数据,本文选择 XGBoost 模型作为公式(1)中的 f 来解决问题,其中模型是通过公式(2)构建的。
3.3 融合过程
由于每个子问题的建模过程都是独立的,所以不能简单地使用从每个子问题评估的监管关系的可信度进行全局排名。因此,本文采用基于 L2 范数的规范化方法来解决这个问题,并且每个子问题的权重 wi,j被规范化为:

其中,p 为每个子问题中基因 j 的候选调控因子的数目。
此外,本文还使用统计方法,通过更新全局权重 wi,j来进一步细化推断 GRN。这种改进是基于这样一个假设:如果一个候选调控因子 i 调控多个靶基因,那么它将是一个重要的调控因子,并且所有调控关系的可信度都应该提高。根据这一点,本文将权重 wi,j更新表述为:



4 结果分析与评估
4.1 数据集与评价指标
本课题的数据集及评价指标均来自于 DREAM 挑战平台(基因调控网络挑战)[18]。该平台为生物学和医学的研究者们提出了众多挑战,并提供了基准数据集来验证评估他们的工作。其中,DREAM5 是第一个利用大规模真实数据集构建 GRNs 的挑战,其中靶基因达到 O(103)数量级、调控基因则为 O(102)。
DREAM5 数据集共包含 4 个网络:网络 1 是通过 in-silico 模拟导出的,另外 3 个网络则是从不同实验中获得的。其中,网络 2 来源于金葡萄杆菌(S.Aureus)相关实验,网络 3 来源于原核生物(E.coli)实验,网络 4 则来源于真核生物(S.cerevisiae)实验。网络 3 和 4 调控关系的金标准来自于 RegulonDB 和 Gene Ontology(GO)两个数据库,而网络 2 没有对应的金标准,故本文暂不考虑。最终选取 DREAM5 挑战的 1、3 和 4 网络数据作为实验数据。
本文选取来自 DREAM5 挑战的静态基因表达数据集,基本信息如表 1 所示。其中,insilico 网络包括 1 643 个靶基因、195 个转录因子(Transcription Factors),金标准中共有 4 012 条调控关系;E.coli 网络包括 4 511 个靶基因、334 个转录因子,金标准中共有 2 066 条调控关系;S.cerevisiae 网络包括 5 950 个靶基因、333 个转录因子,金标准中共有 3 940 条调控关系。
表 1 静态基因表达数据集Table 1 Steady gene expression data set
为评价该方法的性能,本文考虑两种常用的评价指标:接受者操作特征曲线面积(Area Under Receiver Operating Characteristic,AUROC)和正确率-召回率曲线面积(Area Under Precision-Recall Curves,AUPR)。其中,AUROC 是基于真阳率(True Positive Rate,TPR)与假阳率(False Positive Rate,FPR)的接受者操作特征(ROC)范围,AUPR 是根据精确度(Precision)与召回率(Recall)得出的领域。真阳率(TPR)为检测出来的真阳性样本数除以所有真实阳性样本数;假阳率(FPR)为检测出来的假阳性样本数除以所有真实阴性样本数;召回率为真阳性样本在所有检测正确样本总数中的占比。
其中,TP(True Positive)为真阳性的数量;TN(True Negative)为真阴性的数量;FP(False Positive)为假阳性的数量;FN(False Negative)为假阴性的数量。
4.2 随机化和正则化
前人的研究表明,随机化和正则化在重建 GRNs 中是有效的。其中,随机化包括样本抽样和特征选择,如引导程序和子特征。正则化是通过将惩罚项添加到目标函数,进而控制模型的复杂性,回归模型中最常用的正则化技术是 LASSO(L1)和 Ridge(L2)。
本文方法 XGBNet 基于 XGBoost。其中,XGBoost python 软件包提供了用于实现的各种参数,本文选择决策树作为基学习器。参数 max_depth 和 min_child_weight 与模型中每棵树的结构相关,且都设置为 4;将控制每棵树中训练样本比率的参数 subsample 设置为 0.7;参数colsample_bytree 控制每棵树中特征(候选调节器)的比率,并在此处设置为 0.9;学习率 eta 设置为 0.000 8,树的数量设置为 1 000,与大多数“基于树的”方法的默认设置相同。
4.3 实验结果
在 DREAM5 数据集中采用 in-silico 模拟数据、E.coli 和 S.cerevisiae 实验数据来对所提出模型的性能进行评估。更进一步地,选择了几种最新的 GRNs 推断方法,包括 iRafNet[25]、HiDi[24]、PLSNET[22]和 DREAM 挑战赛[18]的获胜者与本文结果进行对比分析,具体结果如表 2 所示。
表 2 为不同方法在 DREAM5 数据集中 3 种网络上的 AUPR 和 AUROC。其中,KO(Knock Out Data)为剔除某个基因后的基因表达数据;SS(Steady-State Expression Data)为平稳基因表达数据。从表 2 可以看出,iRafNet 和 HiDi 都集成了稳态表达数据和剔除数据,而 DREAM5 挑战赛的冠军仅使用稳态表达数据,本方法也仅需用到稳态表达数据,但就网络 1 的 AUPR 和 AUROC 以及网络 3 和 4 的 AUROC 而言,XGBNet 仍然比其他方法具有更好的性能。一个主要原因可能是,由于不完整的 KO 数据所提供的信息很少,对于推断出 GRNs 所起的作用很小。此外,由于生物实验数据的采集误差大、获取途径不一致等问题,导致 5 种方法在网络 3 和 4 的 AUPR 值均较低。
从表 2 数值可看出,本文方法在 in-silico 生成的模拟数据集中,AUPR 和 AUROC 两个评估指标均显著优于现有方法;在 E.coli 和 S.cerevisiae 两种生物的真实实验数据中,AUROC 指标均高于现有最优方法。推测原因在于,本文所提出方法建立特有的集成框架,通过在目标函数中加入 2 种不同的惩罚项,以及限制树结构和剪枝过程,尽可能地避免过拟合,较大幅度地提高了预测准确率;同时用于基因调控网络的多为 bagging 集成方法(以随机森林为典型代表),而以 XGBoost 和 AdaBoost 为主的 Boosting 集成方法性能则通常优于 bagging 集成。两种方法的主要区别在于 bagging 的每个弱学习器都是独立并行学习的,而 Boosting 则顺序地学习这些弱学习器(每个基础模型都依赖于前面的模型),并按照某种确定性的策略将它们组合起来。
4.4 讨论与分析
现阶段对基因静态数据调控网络推断的研究,主要集中在基于信息论、概率图和机器学习的方法中。Zhang 等[10]、Ricardo 等[11]和 Küffner 等[12]使用基因之间的互信息作为度量并以此推导调控网络,来进一步消除间接调控关系的影响,然而仅使用互信息仍不能足够多地提取基因调控的有效信息;Liu 等[14]、Yu 等[15]、Xing 等[16]和 de Campos 等[17]利用叶斯网络学习工具从静态基因数据中计算基因相互作用,从最初的静态贝叶斯到改良的动态贝叶斯,做出了很多的突破,然而效率仍低于现有部分其他方法,同时模型准确率也有待提高;Huynh-Thu 等[19]、Haury 等[20]、Ruyssinck 等[21]和 Guo 等[22]构建基于特征选择框架的集成方法,且用于基因调控网络的多为 bagging 集成方法(以随机森林为典型代表)。与上述基因静态数据调控网络推断研究不同的是,本文选择基于 XGBoost 的集成方法,每个弱学习器都依赖于前一个模型,并按照某种确定性的策略将其组合起来,且着重解决模型过拟合问题,并对集成模型的结果增加归一化和统计学方法处理。当然,本文还存在不足之处,未来工作的重点是进一步研究利用先验信息进行特征选择,并增强方法的可移植性。
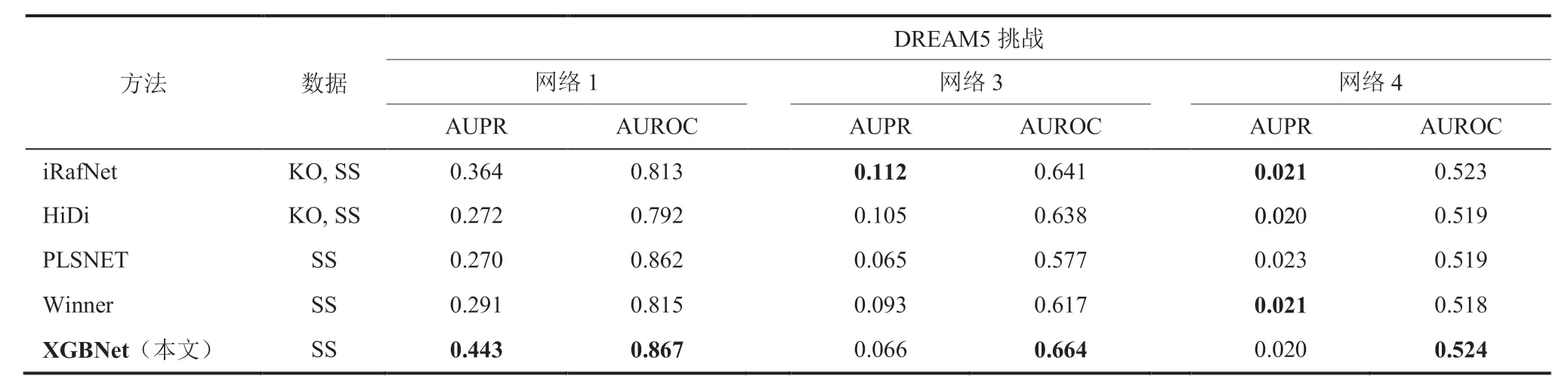
表 2 不同方法结果比较Table 2 Results of different methods
5 结 论
本文提出一种基于 Boosting 集成模型的特征选择框架:针对基因静态数据进行调控网络推断,建立特有的集成框架,同时通过在目标函数中加入 2 种不同的惩罚项,以及限制树结构和剪枝过程,尽可能地避免过拟合,较大幅度地提高了预测准确率。另外,对于不同子问题分别建模得到的权重存在量纲不一致的问题,本文在初始排序基础上增加归一化和统计方法以提高模型准确率。使用来自 DREAM5 挑战的基准数据集测试结果表明,本文所提出的 XGBoost 比现有其他方法获得更好的性能。在未来的研究中,将进一步研究利用先验信息进行特征选择,充分结合多方数据,并增强该方法在其他生物物种基因调控关系应用中的可移植性。
