固相萃取–液相色谱–质谱法测定益母草中7 种真菌毒素
2020-06-03周颖唐登峰李文庭祝清岚马临科昝珂
周颖,唐登峰,李文庭,祝清岚,马临科,昝珂
(1.浙江省食品药品检验研究院,杭州 310053; 2.中国食品药品检定研究院,北京 100050)
益母草来源于唇形科植物益母草的新鲜或干燥地上部分,是临床常用的妇科良药,具有活血调经,利尿消肿,清热解毒的功能[1]。2013 年,国家启动第四次资源普查项目,益母草是中药材资源普查项目的品种之一,鉴于其在采收、储藏、制备及运输过程中因处理不当极易发生霉变,有被真菌毒素污染的可能,而真菌毒素是真菌产生的次级代谢产物,如黄曲霉毒素(AFs)、赭曲霉毒素(OTA)、T–2 毒素(T–2)和展青霉素(Patulin)等,对人体具有较强毒性,因此有必要加强相关真菌毒素的控制[1]。为客观评价益母草药材的质量,并为全国中药资源普查在安全性维度提供数据支持[2],开展益母草中真菌毒素的检测分析势在必行。
近年来,国内外已纷纷开展真菌毒素相应的测定方法及限量要求研究[3–5]。目前文献报道的针对中药材进行真菌毒素检测方法有酶联免疫吸附技 术[6–7]、胶体金免疫层析技术[8–10]、薄层色谱技术[11]、免疫亲和柱净化–高效液相色谱技术[12–14]和高效液相色谱–串联质谱技术[15–18]等。由于中药材复杂基质的干扰,酶联免疫吸附技术在测定真菌毒素时容易产生“假阳性”结果,且受试剂盒差异等条件的影响也较大;胶体金免疫层析技术灵敏度较差,无法精准定量;薄层色谱技术前处理过程使用的大量有机试剂对人体有较大的伤害,同时易造成环境的污染;免疫亲和柱净化–高效液相色谱技术无法检测无荧光和无紫外吸收的真菌毒素;而高效液相色谱–串联质谱技术作为无需衍生化便可检测的分析技术具有分析速度快,灵敏度高、多种组分可同时检测等优势,现已成为较为广泛的真菌毒素检测技术。在益母草中真菌毒素的检测方面,目前国内外对展青霉素、赭曲霉毒素A、黄曲霉毒素G2、黄曲霉毒素G1、黄曲霉毒素B2、黄曲霉毒素B1和T–2 毒素检测分析未见相关报道,本研究将样品中7 种真菌毒素同时提取,采用固相萃取柱富集净化,正离子和负离子切换模式和多反应监测(MRM)模式进行检测,建立了高效液相色谱–串联质谱法同时检测益母草中7 种真菌毒素的方法,以期能为客观评价我国益母草药材的安全性提供技术支持和重要参考。
1 实验部分
1.1 主要仪器与试剂
液相色谱仪:LC–20ADXR 型,日本岛津公司;
三重四极杆串联质谱仪:ABSCIEXQTRAP 5500 型,配有电喷雾离子源,美国应用生物系统公司;
Oasis HLB 固相萃取柱:3 mL,60 mg,美国沃特世公司;
氮气吹干仪:N–EVAPTM111 型,美国Organomation Associates Inc 公司;
Milli-Q 超纯水仪:Milli PAK advantage A10 型,美国密理博公司;
台式大容量冷冻离心机:Centrifuge 5810R 型,德国Eppendorf 公司;
黄曲霉毒素混合对照品溶液:黄曲霉毒素G2(AFG2)、黄曲霉毒素G1(AFG1)、黄曲霉毒素B2(AFB2)、黄曲霉毒素B1(AFB1)的质量浓度分别为0.59,1.18,0.35,1.04 μg/mL,批 号 为610001–20130,中国食品药品检定研究院;
T–2 毒素(T–2):5 mg,纯度为98%,批号为5–MWC–50–1,加拿大TRC 公司;
赭 曲 霉 毒 素A(OTA):10 μg/mL,批 号 为160324,北京曼哈格生物科技有限公司;
展青霉素(Patulin):101.3 μg/mL,5 mL,批号为L15383F,美国Biopure 公司;
乙酸铵:纯度不小于98%,美国西格玛公司;
甲醇:分析纯,国药集团化学试剂有限公司;
乙腈:色谱纯,美国默克公司;
样品:来源于第四次资源普查收集到的82 批次的益母草,涉及全国10 个省、市、自治区,其中有两批为细叶益母草(Leonurus sibiricus L),一批为鬃尾草状益母草(Leonurus chaituroides C.Y.),其余均为其它类益母草(Leonurus japonicus Houtt.);
实验用水为超纯水。
1.2 溶液的制备
标准储备液:分别精密量取黄曲霉毒素混合对照品溶液,加甲醇配制成AFG2,AFG1,AFB2,AFB1质量浓度分别为0.118,0.236,0.07,0.208 μg/mL的混合标准储备液;精密量取适量OTA 加入甲醇配制成0.2 μg/mL 的标准储备液;精密量取展青霉素,加入乙腈配制成6.078 μg/mL 的标准储备液;精密称取T–2 毒素,用乙腈溶解,配制成4.9 μg/mL 的标准储备液。
混合标准储备液:分别精密量取7 种标准储备液于100 mL 容量瓶中,以甲醇溶解,配制成混合标准储备液,于–20℃下保存,浓度见表1。

表1 混合标准储备液
对照品溶液:将混合标准储备液以甲醇稀释2倍后制成对照品溶液。
1.3 仪器工作条件
1.3.1 色谱
色谱柱:ECLIPSE PLUS C18柱(100 mm×2.1 mm,1.7 μm,美国安捷伦科技有限公司);柱温:40 ℃;流速:0.4 mL/min;进样量:5 μL;以5 mmol/L 乙酸铵为流动相A 相,以乙腈为流动相B相,按表2 洗脱条件进行梯度洗脱。

表2 梯度洗脱条件
1.3.2 质谱
电喷雾离子源;正负离子切换扫描模式;电离电压:5 500 V;离子源温度:500℃;气帘气流量:30 L/min;碰撞气流量:5 L/min;多反应监测模式采集;7 种真菌毒素质谱参数及色谱保留时间见表3。
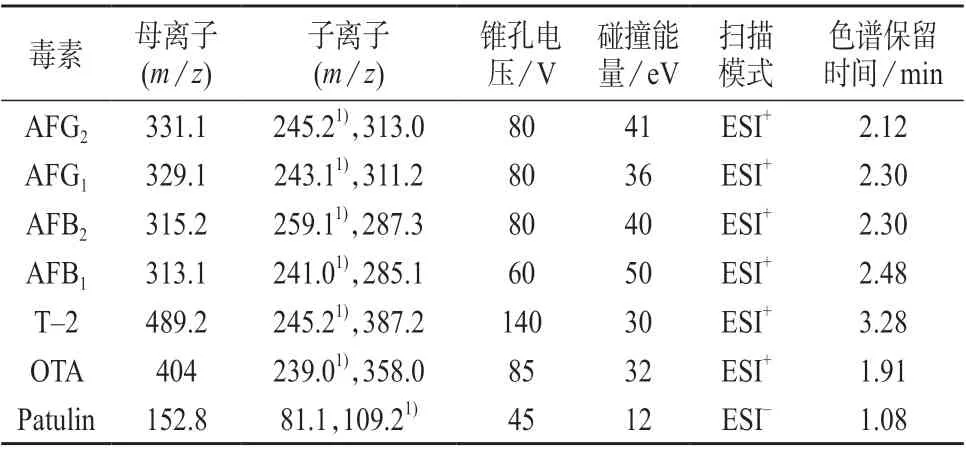
表3 7 种真菌毒素的质谱参数及色谱保留时间
1.4 样品处理
精密称取益母草样品粉末3 g(过二号筛),加入70%甲醇溶液30 mL,超声处理30 min,离心5 min,吸取上清液10 mL,用水稀释至20 mL,摇匀。吸取稀释后的溶液2.5 mL,缓慢通过已经处理好的固相萃取柱(依次用甲醇和水各3 mL 洗脱),收集流出的溶液;然后用2.5 mL 甲醇洗脱,收集甲醇洗脱液,合并之前收集的溶液,用45℃氮气缓慢吹至约1 mL,加入乙腈溶液至2 mL,摇匀,过滤,取续滤液即得待测样品溶液。
2 结果及讨论
2.1 色谱及质谱条件优化
在对质谱系统各参数进行优化过程中,考察了是否含有乙酸铵对优化结果的影响。结果表明,用纯水作为流动相时,OTA 的响应极弱,以致无法确定特征母离子和子离子,用乙酸铵溶液作为流动相时,OTA 的响应信号明显提高,因此最终选择5 mmol/L 乙酸铵溶液–乙腈作为流动相体系。本实验测定的真菌毒素相对较多,为了将残留在色谱柱上化合物充分洗脱,同时缩短分析时间,选用梯度洗脱的方法。
2.2 样品前处理条件优化
在提取方式上,分别考察了超声和均质两种方式。结果表明,超声和均质提取回收率基本相当,故选择简单易行的超声方法进行提取。在定容样品时,分别考察了直接定容至5 mL 和浓缩后定容至2 mL两种提取回收结果,结果表明直接定容至5 mL 响应信号太小,浓缩后定容至2 mL 响应信号显著增大,故选用浓缩后定容的方式。
2.3 线性关系与检出限
精密吸取对照品溶液1,2,3,4,5,6 μL 进样,按1.3 仪器工作条件进行测定。以色谱峰面积为纵坐标,以各组分真菌毒素的质量浓度为横坐标进行线性回归,计算得线性方程和相关系数,以3 倍信噪比对应的浓度作为方法检出限,结果见表4。由表4可知,7 种真菌毒素的质量浓度与色谱峰面积具有良好的线性关系。
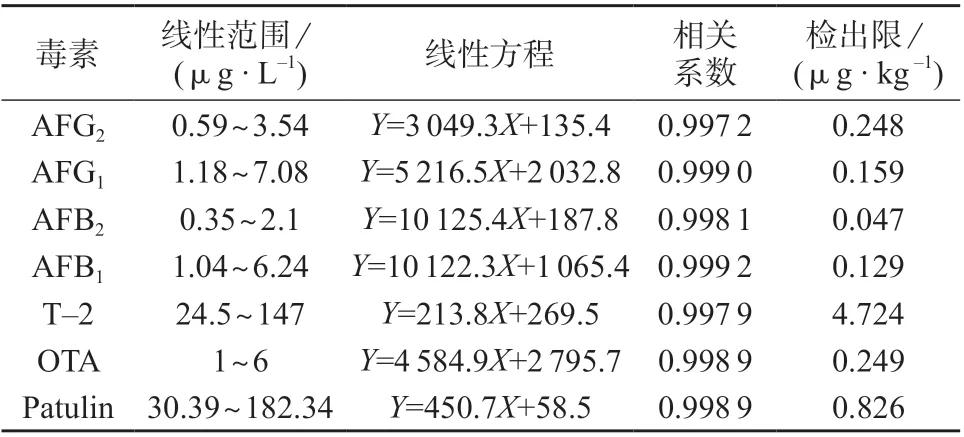
表4 线性范围、线性方程、相关系数、检测限
2.4 加标回收与精密度试验
取益母草阴性样品粉末约3 g(过二号筛),精密称定,加入70%甲醇溶液和混合对照品溶液各15 mL,按照1.4 进行处理,在1.3 条件下测定,计算回收率,结果见表5。由表5 可知,7 种真菌毒素的回收率在72.8%~95.5%之间,相对标准偏差为3.1%~5.7%。7 种真菌毒素的TIC 图见图1,7 种真菌毒素的MRM 色谱图见图2。
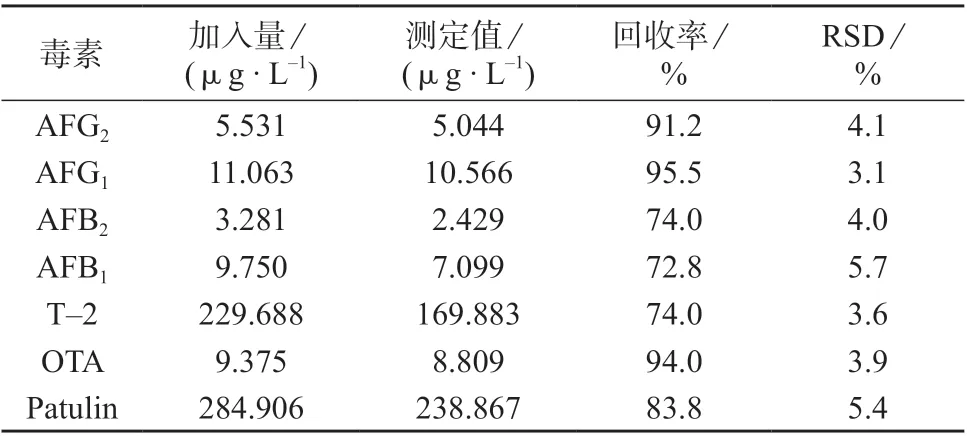
表5 加标回收与精密度试验结果(n=6)
2.5 样品测定
对收集到的82 批益母草样品按本方法进行测定,有3 批益母草样品中检出OTA,分别为1.6,1.2,3.9 μg/kg,其它6 种真菌毒素未见检出。
3 结语
采用固相萃取柱富集净化,以液相色谱–质谱联用方法同时检测益母草中7 种真菌毒素。该方法高效、快速、准确,可满足益母草中真菌毒素的大通量快速筛查和确证。
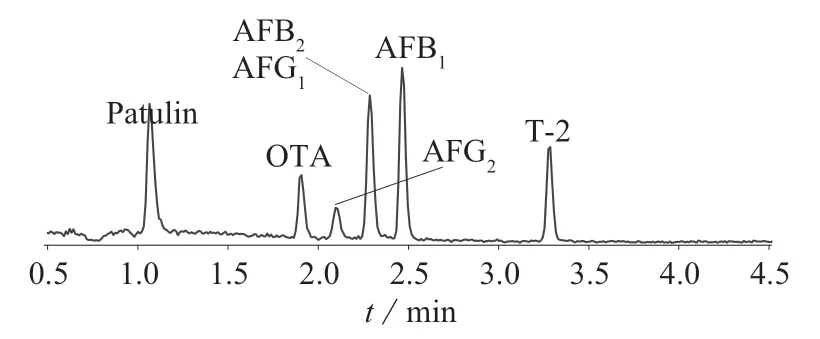
图1 7 种真菌毒素的TIC 图

图2 7 种真菌毒素的MRM 色谱图
