东方蜜蜂微孢子虫侵染中华蜜蜂过程中的高表达基因表达谱及其潜在作用
2020-06-03熊翠玲陈华枝耿四海周倪红周丁丁祝智威陈大福郑燕珍徐国钧
熊翠玲, 陈华枝, 耿四海, 周倪红, 周丁丁, 祝智威, 陈大福,郑燕珍, 徐国钧, 张 曦, 郭 睿
(1. 福建农林大学 动物科学学院(蜂学学院), 福州 350002; 2. 枣庄职业学院, 枣庄 277100)
1 引 言
微孢子虫是一种真菌病原,广泛感染人类、哺乳类、两栖类和昆虫等[1-3].东方蜜蜂微孢子虫(Nosema ceranae)专性寄生成年蜜蜂的中肠上皮细胞,由Fries等人于1996年首次在中华蜜蜂(Apis cerana cerana,简称中蜂)中发现并报道[4],随后该病原迅速传播蔓延至美国和欧洲地区,目前已扩散到世界各地的西方蜜蜂(Apis mellifera)蜂群[5].被N. ceranae感染的蜜蜂寿命减少1/3[6],产蜜量降低40%,分泌蜂蜡量减少25%[7],并伴随有早熟行为[8].此外,N. ceranae还被认为是诱发蜂群崩溃综合症的主要病原之一[9].N. ceranae主要通过粪-口或口-口等途径在蜜蜂个体间传播,当孢子进入蜜蜂中肠,在碱性环境的刺激下,孢子发芽并弹射出高度压缩的极丝,极丝穿入上皮细胞并将有感染性的孢质原浆注入其中;N. ceranae经过增殖形成孢子体,最终导致宿主细胞裂解,释放出的成熟孢子继而感染临近的细胞,或者通过粪便排泄到外界环境中,侵染其他蜜蜂个体[10].
前人对于N. ceranae的研究集中在病原鉴定、流行病学、病理学、侵染机理等方面[10-12].Cornman等人于2009年完成N. ceranae的基因组组装并公布其序列及注释信息,为其组学及分子生物学研究提供了基础.较之蜜蜂,N. ceranae的组学研究进展缓慢,相关信息较为缺乏.Huang[13]等通过对N. ceranae感染1-6d的A. mellifera中肠进行转录组测序和分析,发现在感染过程中宿主的细胞凋亡基因表达受到抑制,N. ceranae在一个繁殖周期中有1 122个基因差异表达,并分为4种表达模式.Evans等发现1 545个A. mellifera基因可能受到N. ceranae的5个miRNA的调控,这些miRNA聚集成6个共表达组,6个组中的4个(918个基因)与至少1个miRNA的表达显著相关,N. ceranae的miRNA倾向于与病原基因正相关而与宿主基因负相关,表明N. ceranae的miRNA可调控自身基因表达也能跨界调控宿主的部分基因表达[14].目前,前人仅对侵染A. mellifera的N. ceranae进行了一些组学研究,而对于侵染东方蜜蜂(Apis cerana)的N. ceranae以及二者互作,还缺乏相关研究报道.近期,笔者团队结合链特异性建库方法和RNA-seq技术对N. ceranae孢子进行了全转录组测序,通过生物信息学方法分别预测出83个长链非编码RNA(long non-coding RNA, lncRNA)和204个环状RNA(circular RNA, circRNA),并对它们的数量、种类、结构特征、表达谱和调控网络进行了系统的分析和验证[15-16].
A. cerana是N. ceranae的原始寄主,二者经过漫长的协调进化已相互适应.有研究表明N. ceranae对A. mellifera具有更强的毒力[5].因此,对侵染A. cerana的N. ceranae进行组学研究,可为解析N. ceranae的侵染机制和病原-宿主的适应性进化机制提供有价值的信息和线索.为了在转录组水平探究在N. ceranae侵染中蜂过程中的的作用,本研究利用基于链特异性建库方法的RNA-seq技术对N. ceranae侵染7 d和10 d的中蜂工蜂中肠进行测序,筛选出病原的高表达基因(highly expressed gene, HEG),并通过生物信息方法对N. ceranae的HEG进行深入分析,并通过RT-PCR技术对部分HEG进行验证,以期揭示HEG在N. ceranae侵染中蜂过程的作用,为深入N. ceranae的侵染机制提供有益的信息和线索.
2 材料与方法
2.1 生物材料
本研究中使用的中蜂取自福建农林大学动物科学学院(蜂学学院)教学蜂场;N. ceranae孢子[15]由福建农林大学蜂学学院蜜蜂保护课题组纯化和保存.
2.2 方 法
2.2.1 蜂群选择 挑选外观健康、群势较强的10群中蜂,在每个蜂箱的巢房门口随机抓取6只工蜂,小心拉取中肠,加适量无菌水后充分研磨后取少许研磨液进行显微制片和镜检,利用前人报道的N. ceranae特异性引物(F: CGGCGACGATGTGATATGAAAATATTAA, R: CCCGGTCATTCTCAAACAAAAAACCG)[17]和N. apis特异性引物(F: GGGGGCATGTCTTTGACGTACTATGTA, R: GGGGGGCGTTTAAAATGTGAAACAACTATG)[17]对镜检不到孢子的样品进行PCR检测,将镜检和PCR检查都为阴性的中蜂蜂群作为本研究的实验蜂群.
2.2.2 中蜂的饲喂感染及人工饲养 参照郭睿[17]等的方法进行中蜂的饲喂接种及人工饲养,简述如下:将实验蜂群内封盖子较多的巢脾迅速提至实验室,放入34±0.5 ℃恒温培养箱,将刚出房的工蜂(当天记为0 d)放进干净的塑料盒(塑料盒四周打孔通风,30只/盒),每个盒子上方中心插入一支装有50%(w/v)无菌糖水的饲喂器;(3)34±0.5 ℃培养24 h,将上述工蜂饥饿处理2 h,然后给每只工蜂饲喂5 μL糖水(含1×106个N. ceranae孢子),舍弃未食尽糖水的工蜂,将食尽糖水的工蜂用于后续实验;(4)每日检查工蜂的存活情况,及时清理死亡的工蜂,每隔24 h更换糖水.本实验进行三次生物学重复.
2.2.3 测序样品准备及二代测序 因蜜蜂中肠是N. ceranae寄生和二者互作的主要部位,故本研究选取N. ceranae侵染的中蜂工蜂中肠作为测序材料.分别拉取N. ceranae感染7 d(即8日龄)和10 d(即11日龄)的工蜂中肠,迅速移至RNA free的EP管中,三只中肠并入一管,液氮速冻后转移保存于-80 ℃超低温冰箱中备用.将侵染中蜂8日龄和11日龄工蜂的N. ceranae分别设为Nc1和Nc2.Nc1组的3个生物学重复为:Nc1-1、Nc1-2和Nc1-3;Nc2组的3个生物学重复为:Nc2-1、Nc2-2、Nc2-3.利用RNAiso Reagent试剂盒(TaKaRa公司,中国)抽提上述样品的总RNA,再用RNase-free DNase I去除上述样品中基因组DNA残留,然后用琼脂糖凝胶电泳进行片段大小选择.参照郭睿等[18]的方法构建cDNA文库.建好的文库委托广州基迪奥生物技术有限公司进行双端测序,测序平台为Illumina HiSeqTM4000.原始数据已上传NCBI的SRA数据库,BioProject号:PRJNA562784.
2.2.4 数据质控及相关生物信息学分析 对于获得的原始数据读段(raw reads),利用Perl脚本去除有adaptors、未知核苷酸比例大于5%和低质量的reads,获得有效读段(clean reads).按照下述步骤获得纯净的N. ceranae数据:(1)用短reads比对工具bowtie将clean reads比对到核糖体数据库(最多允许错配5个);(2)利用TopHat2软件将未比对上的数据比对A. cerana的参考基因组(assembly ACSNU-2.0)以去除宿主本身的clean reads;(3)进而利用TopHat2软件将未比对上剩余的clean reads继续比对到N. ceranae的参考基因组(assembly ASM98816v1),比对上的clean reads即为病原的数据.进一步再利用Cufflinks软件对比对上的clean reads进行组装.
各组基因的表达量用公式FPKM =Total exon fragment/[Mapped fragment (millions) × exon length (KB)]计算.利用R包计算个样品之间的相关性系数.利用OmicShare在线分析工具(http://www.omicshare.com/tools/index.php/Home/Index/index.html)对各个样品的HEG(FPKM >15)进行Venn分析,以及对共有HEG进行GO分类和KEGG代谢通路(pathway)富集分析等生物信息学相关分析.
2.2.5 HEG的RT-PCR验证 为证明测序数据的可靠性,从共有HEG中随机选择8个进行RT-PCR验证.根据所选HEG的序列信息,使用Primer Premier 5软件设计特异性引物,委托上海生工生物工程有限公司进行引物合成,相关引物序列信息详见表1.利用AxyPrepTMMultisource Total RNA Miniprep试剂盒(AXYGEN公司,中国)分别抽提上述受N. ceranae感染的中蜂工蜂中肠样品的总RNA,作为模板进行反转录,得到的cDNA作为模板进行PCR扩增.PCR反应体系为20 μL,包括PCR Mixture 10 μL,正、反向引物各1 μL,cDNA模板1 μL,无菌水7 μL.PCR反应条件为:94 ℃预变性5 min;94 ℃50 s,58 ℃退火30 s,72 ℃延伸1 min,共35个循环;72 ℃延伸10 min.PCR扩增产物经1.5%琼脂糖凝胶电泳检测.

表1 RT-PCR的引物信息
3 结果与分析
3.1 测序数据概览
比对上A. cerana基因组的clean reads数分别为180 237 114和36 054 033条,Q20和Q30分别在98.41%和95.28%及以上(表2).此外,各组内各生物学重复之间的Pearson相关性均在0.969 1及以上(图1).以上结果说明本研究中的测序样品重复性较好,高通量测序数据质量好,可满足本研究中N. ceranae的HEG分析.
表2 测序数据的信息统计
Tab.2 Summary of sequencing data information

样本Samples比对上N. ceranae基因组的有效读段Clean reads mapped to the N. ceranae genome99%碱基正确率/%Q20/%99.9%碱基正确率/%Q30/%Nc1-138 984 05898.7596.30Nc1-235 848 94498.7496.23Nc1-3105 404 11298.6095.85Nc2-17 568 77498.4195.28Nc2-213 016 30398.8096.42Nc2-315 468 95698.6696.02
3.2 HEG的筛选及Venn分析
从Nc1和Nc2中分别筛选出1 482和901个HEG.共有HEG为890(59.6%)个,特有HEG分别为592(39.7%)和11(0.7%)个(图2).Nc1和Nc2的部分特有HEG信息详见表3和表4.

图1 Nc1和Nc2中不同生物学重复间的相关性Fig.1 Pearson correlations between every two biological repeats within Nc1 and Nc2 groups

图2 Nc1和Nc2中HEG的Venn分析
Fig.2 Venn analysis of HEGs in Nc1 and Nc2 groups
3.3 共有及特有HEG的GO分类
GO分类结果显示分别有298、302和175个共有HEG富集在生物学进程、细胞组分和分子功能,共涉及24个功能条目,富集前5位分别是代谢进程(230个HEG)、催化活性(213个HEG)、结合(200个HEG)、细胞进程(198个HEG)、细胞(139个HEG)(图3).
Nc1的特有HEG涉及21个功能条目,富集前10位分别是代谢进程(67个HEG)、细胞进程(64个HEG)、催化活性(63个HEG)、结合(61个HEG)、单组织进程(32个HEG).Nc2的特有HEG涉及4个功能条目,分别为代谢进程(1个HEG)、细胞进程(1个HEG)、单组织进程(1个HEG)和催化活性(1个HEG).

表3 Nc1的前11位特有HEGs
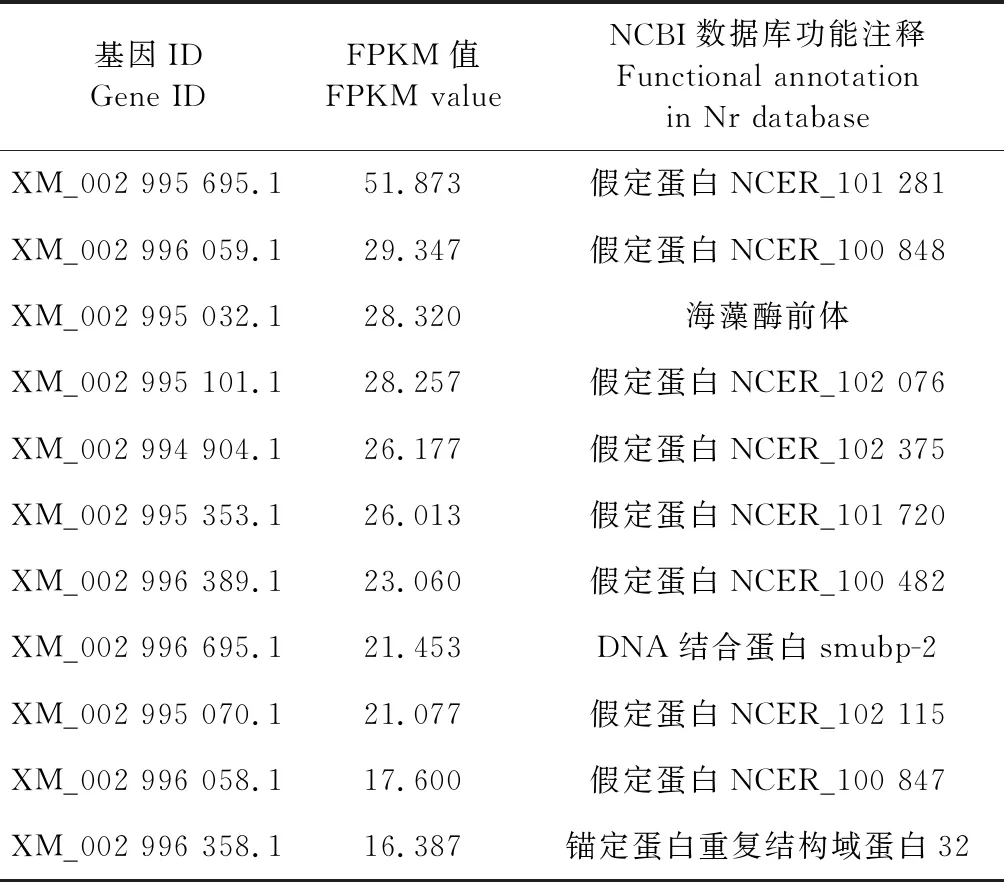
表4 Nc2的特有HEGs

图3 Nc1和Nc2的共有HEGs的GO分类Fig.3 GO categorization of shared HEGs in Nc1and Nc2 groups
3.4 共有及特有HEG的KEGG代谢通路富集分析
KEGG代谢通路富集分析结果显示,共有HEG富集在新陈代谢、遗传信息加工、环境信息加工、细胞组分、有机体系统等5大类的72条代谢通路,其中富集前10位分别是核糖体(53个HEG)、内质网蛋白加工(33个HEG)、蛋白酶体(27个HEG)、真核生物的核糖体生物合成(24个HEG)、嘧啶代谢(23个HEG)、RNA转运(21个HEG)、嘌呤代谢(21个HEG)(图4).进一步分析发现2个共有HEG富集在MAPK信号通路(图5).
Nc1的特有HEG富集在51条代谢通路,富集前10位分别是泛素介导的蛋白水解(12个HEG)、RNA转运(11个HEG)、细胞周期-酵母(11个HEG)、嘌呤代谢(11个HEG)、真核生物的核糖体生物合成(11个HEG).Nc2的特有HEG仅富集在1条代谢通路,即淀粉和蔗糖代谢.

图4 Nc1和Nc2的共有HEG的KEGG代谢通路分析Fig.4 KEGG pathway enrichment analysis of shared HEGs in Nc1 and Nc2 groups
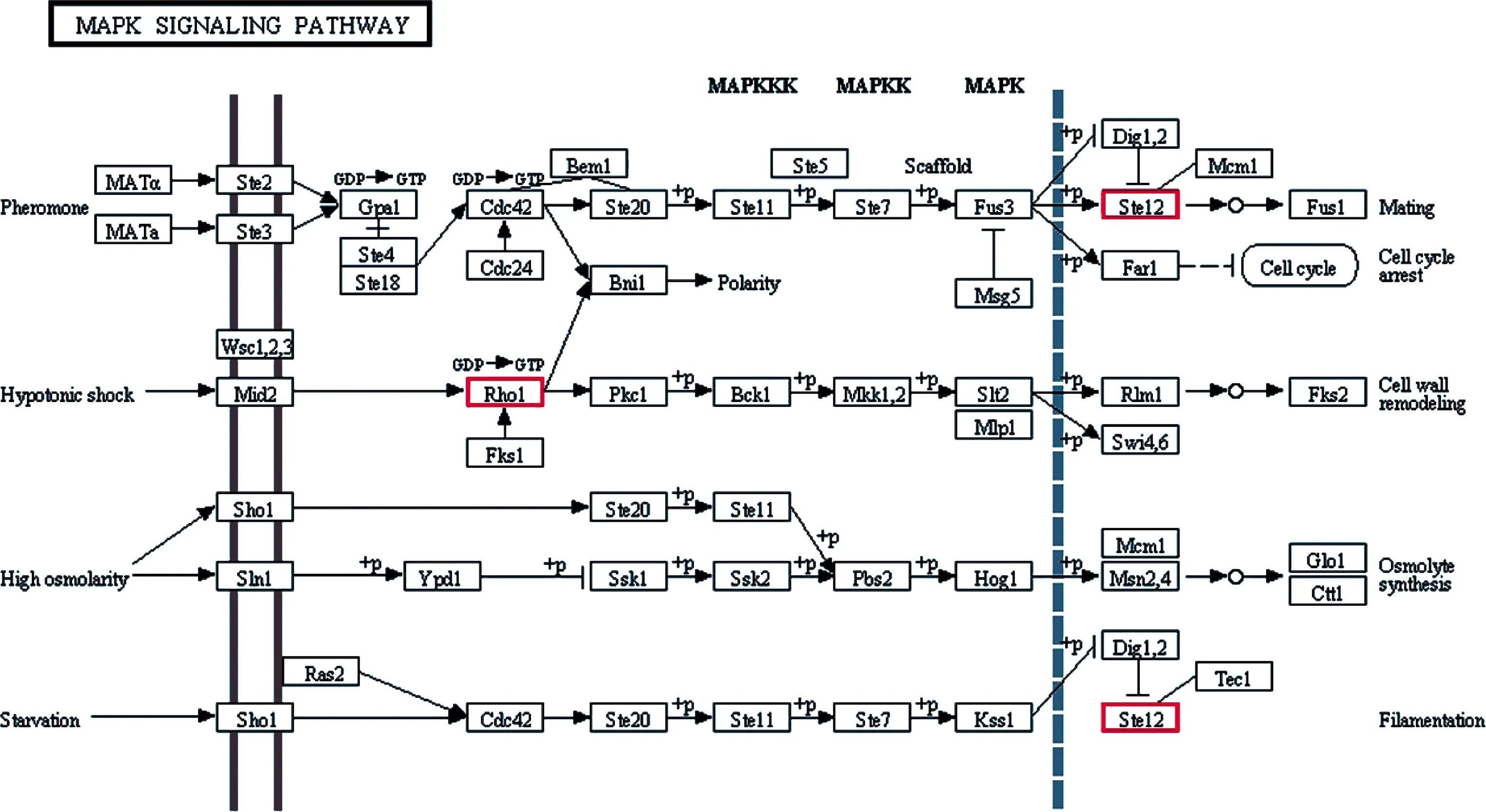
图5 N. ceranae的MAPK信号通路概貌红框表示Nc1和Nc2的共有HEF.Fig.5 Overview map of MAPK signaling pathway in N. ceranaeRed boxes indicate the shared HEF between Nc1 and Nc2.

图6 8个共有HEG的RT-PCR验证 A:磷酸甘露糖酶编码基因; B: 假设蛋白NCER_102326编码基因; C: RNA聚合酶II CTD磷酸酶编码基因; D: 水通道样蛋白编码基因; E: 26s蛋白酶体调节亚基4编码基因; F: 蛋白酶体亚基-1型编码基因; G: 蓖麻毒素b凝集素编码基因; H: 鸟苷酸结合蛋白sar1编码基因Fig. 6 RT-PCR verification of eight shared HEGsA:phosphomannomutase encoded gene; B: hypothetical protein NCER_102326 encoded gene; C: RNA polymerase ii ctd phosphatase encoded gene; D: aquaporin-like protein encoded gene; E: 26s proteasome regulatory subunit 4 encoded gene; F: proteasome subunit beta type-1 encoded gene; G: ricin b lectin encoded gene; H: gtp-binding protein sar1 encoded gene
3.5 HEG的RT-PCR验证
电泳结果显示8个随机挑选的共有HEG在Nc1和Nc2中均扩增出目的条带(图6),初步证实了本研究预测的HEG真实存在.
4 讨论与结论
N.ceranae广泛感染世界各地的蜂群,除能对蜜蜂造成能量胁迫和免疫抑制等影响外,还能与其他生物或非生物胁迫因子共同危害蜜蜂健康[6-8].此前的组学相关研究主要集中在N. ceranae与A. mellifera的互作方面,有关N. ceranae与A. cerana互作的组学研究鲜有报道.为探究N. ceranae在侵染中蜂过程的HEG表达谱及功能,本研究利用RNA-seq技术对N. ceranae侵染7 d和10 d的中蜂工蜂中肠进行测序,得到高质量的测序数据,并通过连续比对A. cerana和N. ceranae的基因组获得病原本身的转录组数据.从Nc1和Nc2中分别筛选出1 482和901个HEG,其中有890个在二者中皆高量表达(FPKM分别介于72 073.050-15.76和75 951.357-15.737),分别有592个HEG(FPKM介于2 962.700-15.003)和11个HEG(FPKM介于51.873-16.387)特异性高量表达.推测上述共有HEG在N. ceranae的侵染过程发挥基础性作用,而特有HEG在侵染的不同阶段具有特定功能,但具体的功能有待进一步研究.Cornman等曾利用罗氏454平台对N. ceranae孢子进行测序,通过相关生物信息学软件对基因组进行了组装和注释[19].然而,由于缺失基因功能研究技术体系,包括N. ceranae在内的微孢子虫的转基因操作技术尚未完全建立,仅在家蚕微孢子虫中有过一例转基因研究报道[20],因而,包括N. ceranae在内的微孢子虫的基因功能研究近乎停滞,加上N. ceranae的组学数据相对有限,导致其基因功能注释信息较不完善.本研究发现,Nc1和Nc2的HEG共包含362个假定蛋白编码基因,这些HEG在各大主流蛋白功能数据库尚缺乏功能注释信息,其完善需要更多的组学数据的支持以及转基因操作技术体系的建立.
本研究中,Nc1和Nc2的共有HEG涉及24个功能条目;分别有139、139、64、34、30和17个HEG富集在细胞、细胞组件、细胞器、细胞膜、细胞膜组件和细胞器组件,另有198和27个HEG富集在细胞进程和细胞组分组织或生物合成,表明N. ceranae在侵染中蜂的过程中伴随着活跃的细胞生命活动,以促进自身在宿主细胞内的增殖.此外,上述共有HEG参与了72条代谢通路,包含碳代谢(15)和糖酵解/糖异生(11)等41条物质代谢通路,氧化磷酸化(8)和磷酸戊糖途径(5)2条能量代谢通路.上述结果表明N. ceranae在侵染过程中的物质和能量代谢较为旺盛.Ponton等[21]的发现N. ceranae侵染A. mellifera后即利用宿主的物质和能量进行自身遗传物质的复制、转录和翻译.本研究发现分别有23、21和21个N. ceranae的HEG富集在嘧啶代谢、RNA转运和嘌呤代谢;此外,分别有53、33和19个HEG富集在核糖体、内质网蛋白质加工和氨酰基-tRNA生物合成.这些结果表明N. ceranae在中蜂工蜂中肠上皮细胞内的增殖过程中同时进行着较为活跃的DNA复制、RNA转录和蛋白质翻译等活动.
信号通路指能将细胞外的信号经细胞膜传入细胞内的一系列酶促反应通路,真菌通过MAPK等信号通路转导系统对外界环境的变化作出相应的应答,进而对生长和毒力等相关基因的表达进行调控,从而影响孢子形成、营养感知和形态建成等生物学过程[22].陈大福等[23]发现对于侵染中蜂6日龄幼虫的蜜蜂球囊菌(Ascopshaera apis,简称球囊菌),富集在MAPK信号通路的HEG远多于体外培养的球囊菌,推测MAPK信号通路在球囊菌的侵染过程作用关键.本研究中,2个共有HEG富集在MAPK信号通路,暗示此信号通路在N. ceranae的侵染过程中发挥重要作用.Jaroenlak等[24]在虾肝肠微孢子虫(Enterocytozoon hepatopenaei)中发现一种分布在内生孢子和外孢子层的新孢壁蛋白(EhSWP1),作为病原在侵染早期的重要毒力因子,可能通过结合肝素附着到宿主细胞表面.极管蛋白与N. ceranae毒力大小有关,有研究表明干扰N. ceranae的极性管蛋白3基因(ptp3)可抑制N. ceranae增殖[25].Paldi等发现宿主摄入靶向N. ceranae的ADP/ATP转移蛋白基因的siRNA能导致特定的基因沉默,进而会影响N. ceranae感染水平和宿主的生理状况[26].本研究中,孢壁蛋白基因(XM_002996303.1, Nc1中FPKM=24 434.550, Nc2中FPKM=33 275.467)、极管蛋白基因(XM_002995446.1, Nc1中FPKM=17 490.823, Nc2中FPKM=29 944.273)、蓖麻毒素B链基因(XM_002995387.1, Nc1中FPKM=698.757, Nc2中FPKM=1 547.050)、ABC转运体基因(XM_002995069.1,Nc1中FPKM=133.697, Nc2中FPKM=265.673)、ATP/ADP转移酶基因(XM_002995682.1,Nc1中FPKM=10 832.593, Nc2中FPKM=7 752.717)和丝氨酸苏氨酸蛋白激酶基因(XM_002995071.1, Nc1中FPKM=337.033, Nc2中FPKM=266.083)等毒力基因均为高量表达,表明N. ceranae在其侵染中蜂工蜂的过程中通过维持上述毒力基因的高表达水平以提升毒力蛋白的合成代谢,进而促进自身在宿主细胞内的增殖.海藻糖广泛存在于酵母等真菌的孢子和子实体中,在脱水、干旱、高温、冷冻、高渗透压和有毒试剂等不良环境的胁迫下起到保护作用[27].海藻糖与海藻糖酶结合会产生大量葡萄糖,导致细胞内渗透压升高促进微孢子虫对宿主的侵染,在微孢子虫孢子的发芽过程中海藻糖酶催化海藻糖水解为葡萄糖为孢子正常发育提供能量[28].本研究发现,海藻糖磷酸合酶1基因(XM_002996572.1, Nc1中FPKM=239.363, Nc2中FPKM=301.7)和海藻糖-6-磷酸合酶基因(XM_002996577.1, Nc1中FPKM=73.413, Nc2中FPKM=200.397)在Nc1和Nc2中皆高量表达,推测N. ceranae通过调控海藻糖磷酸合酶合成速率为自身增殖提供能量.笔者团队在前期研究中发现对于N. ceranae侵染7d和10d的中蜂工蜂中肠,随着侵染时间的延长,与宿主物质和能量代谢相关的HEG数量持续降低[29],表明宿主的物质和能量代谢受到N. ceranae的抑制,体现出N. ceranae通过与中蜂互作而对后者的部分生命活动进行操纵.前人研究发现通过RNA干扰(RNAi)对N. ceranae的ATP/ADP转移酶和裸角质层(Naked cuticle)等毒力因子基因进行沉默的效果良好[26, 30].未来可针对N. ceranae的海藻糖磷酸合酶1基因和海藻糖-6-磷酸合酶基因设计特异性的小干扰RNA(siRNA),通过注射或饲喂感染蜜蜂对该上述病原基因进行沉默,有望用于蜜蜂微孢子虫病的治疗.值得一提的是,本研究发现若干表达水平相对特别高的基因,如部分SR22蛋白基因(XM_002996758.1, Nc1中FPKM=61 410.717, Nc2中FPKM=75 951.357)、假定蛋白NCER_100570基因(XM_002996307.1, Nc1中FPKM=69 904.740, Nc2中FPKM=75 053.357)和延伸因子1基因(XM_002995284.1, Nc1中FPKM=33 358.027, Nc2中FPKM=34 716.303),下一步拟通过RT-PCR和RACE技术得到上述HEG的cDNA全长序列,进而在核酸和蛋白水平开展相关的功能研究.
综上所述,N. ceranae可能通过MAPK信号通路对中蜂中肠的碱性环境作出应答,促进孢壁蛋白、极管蛋白、蓖麻毒素B链、ABC转运体、ADP/ATP转移蛋白和丝氨酸苏氨酸蛋白激酶等毒力因子基因的高量表达,刺激孢子萌发和菌丝弹射,促进N. ceranae对中蜂中肠上皮细胞的侵染;在中蜂中肠细胞内,激活ADP/ATP转移蛋白和海藻糖磷酸合酶基因的表达,对宿主造成能量胁迫,为自身增殖提供能量.本研究通过深入分析侵染中蜂工蜂的N. ceranae的HEG表达谱,揭示了HEG在病原侵染过程中的潜在作用,揭示N. ceranae通过维持孢壁蛋白、极管蛋白、蓖麻毒素B链、ABC转运体、ADP/ATP转移蛋白和丝氨酸苏氨酸蛋白激酶等毒力因子基因,以及ATP/ADP转移酶、海藻糖磷酸合酶1和海藻糖-6-磷酸合酶等能量代谢相关蛋白编码基因的高量表达,促进自身在中蜂工蜂中肠的侵染过程.研究结果为明确N. ceranae对中蜂的侵染机制提供了有益信息和线索.
