己酸乙酯/月桂酸乙酯及其二元混合物界面性质的分子动力学模拟
2020-06-03刘晓娜赵贯甲尹建国马素霞
刘晓娜,赵贯甲,尹建国,马素霞
(太原理工大学 a.电力学院,b.循环流化床高效清洁与利用山西省重点实验室,太原 030024)
随着我国经济的发展,人民生活水平不断提高,汽车工业也获得了快速的发展,汽车保有量剧增,与此同时汽车尾气排放已经成为城市雾霾的主要来源之一。开发新型清洁燃料及其添加剂作为从排放源头控制污染的重要手段已经成为政府、学界和社会各界的共同认识。生物柴油是一种含氧燃料,具有较高的十六烷值,单独或与化石柴油混合作为调和油使用时,可以有效提高燃烧效率,并且显著降低污染物的排放。生物柴油的来源广泛,可以从植物油、动物油、微生物油、废弃油脂等通过酯化反应获得,其组分一般为碳原子数为8—18的脂肪酸甲酯或脂肪酸乙酯[1-2]。研究表明,与普通化石柴油相比,纯生物柴油燃烧时除NOx随负荷增加略有增大外,CO,HC和碳烟等有害颗粒物排放浓度可分别降低29.0%,24.9%和43.1%,与化石柴油掺混燃烧时,污染物排放量随生物柴油比例增加呈减小趋势[3]。己酸乙酯和月桂酸乙酯是生物柴油的组分,常用作化石柴油的添加剂,其纯质和二元混合物可以作为模型燃料,用以研究生物柴油的喷射雾化、燃烧和排放等特性[4-5]。
燃油理化性质是设计内燃机喷射系统的基础数据,譬如表面张力,是影响燃料雾化液滴粒径的关键参数之一[6],一般具有较高表面张力的燃油更难以破碎成较小液滴,增加了燃油的贯穿距离,并降低了燃烧效率,导致污染物的排放量增加[7]。实验测量是精确获取燃油理化性质的主要手段,但受限于实验条件,获得的数据往往集中于有限的温度、组分和压力范围,且难以获取界面层厚度和密度分布等气-液界面的微观性质[8],这些性质有助于从微观视角对燃烧进行分析和诊断,也为新型燃料和燃料添加剂的开发提供了支持。分子动力学模拟既可以提供宏观的表面张力信息,也可以获取微观的界面层分布特性,可以作为实验测量的补充手段,深入分析复杂燃油体系的界面信息。本课题组采用分子动力学模拟方法,研究了293~463 K温度范围内,不同配比的正十二烷与辛酸甲酯混合物的密度,并指出opls-aa力场参数较适合脂肪酸酯类密度的模拟[9]。本文继续使用分子动力学模拟的方法,通过opls-aa力场参数研究己酸乙酯、月桂酸乙酯及其在不同配比下的二元混合物的表面张力与密度,并且从微观层面上分析不同体系的密度分布以及界面层厚度的变化规律。本文工作不仅为燃料喷射系统设计和新型燃料的开发提供参考数据,同时也验证opls-aa力场进一步应用于脂肪酸酯类界面性质模拟的可靠性,以便扩宽该力场适用范围,获取此类物质在更宽的温度、压力和组分范围内与其喷射特性相关的界面性质,为其燃烧分析和诊断提供支撑。
1 模拟方法与细节
1.1 模拟方法
进行分子动力学模拟,首先需要建立分子拓扑结构,选择力场参数,设定所有粒子的坐标和速度等初始信息,然后根据提供的信息计算每个粒子的受力,进而利用牛顿运动方程不断更新构型,遍历整个体系,最终获得所有粒子的运动轨迹以及能量、压力和温度等信息。
采用Irving和Kirkwood[10]提出的方法表示压力张量,则体系的表面张力可以表示为:
γ=0.5Lz
(1)
式中:γ为表面张力,mN/m;Lz为盒子的Z方向的尺寸,nm;Pxx、Pyy和Pzz为体系在X、Y和Z方向的压力,MPa.
1.2 模拟细节
模拟使用的工作站配置为: 显卡型号为GeForce RTX 2080 Ti,11 GB显存,核心数为4 352个;CPU为8核Intel至强 E5-2609 v4处理器,最大处理速度为1.70 GHz;操作系统为Centos,64位版本为7.6.模拟软件采用GPU加速版Gromacs,版本为2016.4.模拟采用opls-aa全原子力场,初始构型为6 nm×6 nm×6 nm的面心立方体盒子,具体的粒子数设定如表1所示。模拟分为三个阶段,第一阶段为能量最小化,采用最速下降搜索方法,积分步长为0.02 ps.为消除边界效应,将体系在X、Y和Z方向设为周期性边界条件,且认为最大能量小于1.0 kJ/mol时收敛。第二阶段为平衡阶段,选择NPT系综,设定压力为0.1 MPa,平衡时长为10 ns,获得更紧凑的结构和更稳定的体系。最后是成品模拟阶段,将平衡后的体系盒子增大,使得Lx=Ly≈0.5Lz,体系如图1所示。选择NVT系综,范德华作用力的计算采用cut-off方法,截断半径设为1.4 nm,控温方法为V-rescale,模拟步长为0.002 ps,模拟时间为30 ns.

表1 己酸乙酯和月桂酸乙酯粒子填充数量与摩尔分数比Table 1 Filling amount and mole fraction ratio of ethyl hexanoate and ethyl dodecanoate in different systems
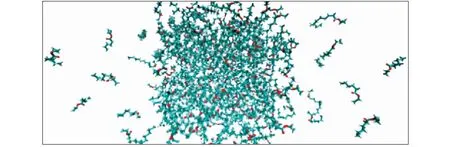
图1 己酸乙酯、月桂酸乙酯摩尔分数比为0.50∶0.50的混合物在398.15 K下的成品模拟构型Fig.1 Final configuration for the mixture of ethyl hexanoate and ethyl dodecanoate with a mole fraction ratio of 0.50∶0.50 at 398.15 K
2 模拟分析
2.1 表面张力模拟数据分析
本文对己酸乙酯、月桂酸乙酯纯质和二者混合物的表面张力进行了研究,纯质和混合物的模拟温度范围分别为298.15~448.15 K和308.15~413.15 K.表2列出了表面张力的模拟值、实验值以及二者的相对偏差数据。将己酸乙酯和月桂酸乙酯的表面张力拟合为van der Waals形式:
σ=σ0(1-Tr)1.26[1+σ1(1-Tr)0.5+
σ2(1-Tr)] .
(2)
式中:σ0(单位mN·m-1),σ1和σ2为拟合参数;Tr为对比温度,Tr=T/Tc;Tc为临界温度,K。己酸乙酯的拟合参数分别为σ0=227.152 35 mN/m,σ1=-2.080 94,σ2=1.557 65;月桂酸乙酯的拟合参数分别为σ0= 143.355 51 mN/m,σ1=-1.917 89,σ2=1.531 57.己酸乙酯模拟值与方程(2)拟合的表面张力值的最大偏差为2.4%,平均绝对偏差为0.8%;月桂酸乙酯模拟值与方程(2)拟合的表面张力值的最大偏差为4.4%,平均绝对偏差为2.0%,两个方程拟合得到的数据与实验比较的平均偏差为7.8%.如图2所示,表面张力的模拟值随温度的升高呈线性减小趋势,变化规律与实验值一致。表面张力主要是由于分子间的相互吸引形成的,可以发现对于纯物质在同一温度下,碳链较长的分子表面张力较大。对于本文的两种物质混合时,混合物的表面张力均介于两种纯质之间,且混合物的表面张力值更接近摩尔分数比例较大的物质。由表2和图3可知己酸乙酯、月桂酸乙酯及其二元混合物表面张力的模拟值与实验值的平均绝对偏差分别为2.9%,5.1%,6.5%.除个别数据点偏差较大外,大部分模拟都准确地重复了实验数据。因此,opls-aa力场不但可以准确预测脂肪酸酯类纯质及其混合体系的密度,同时也可以预测其表面张力性质。
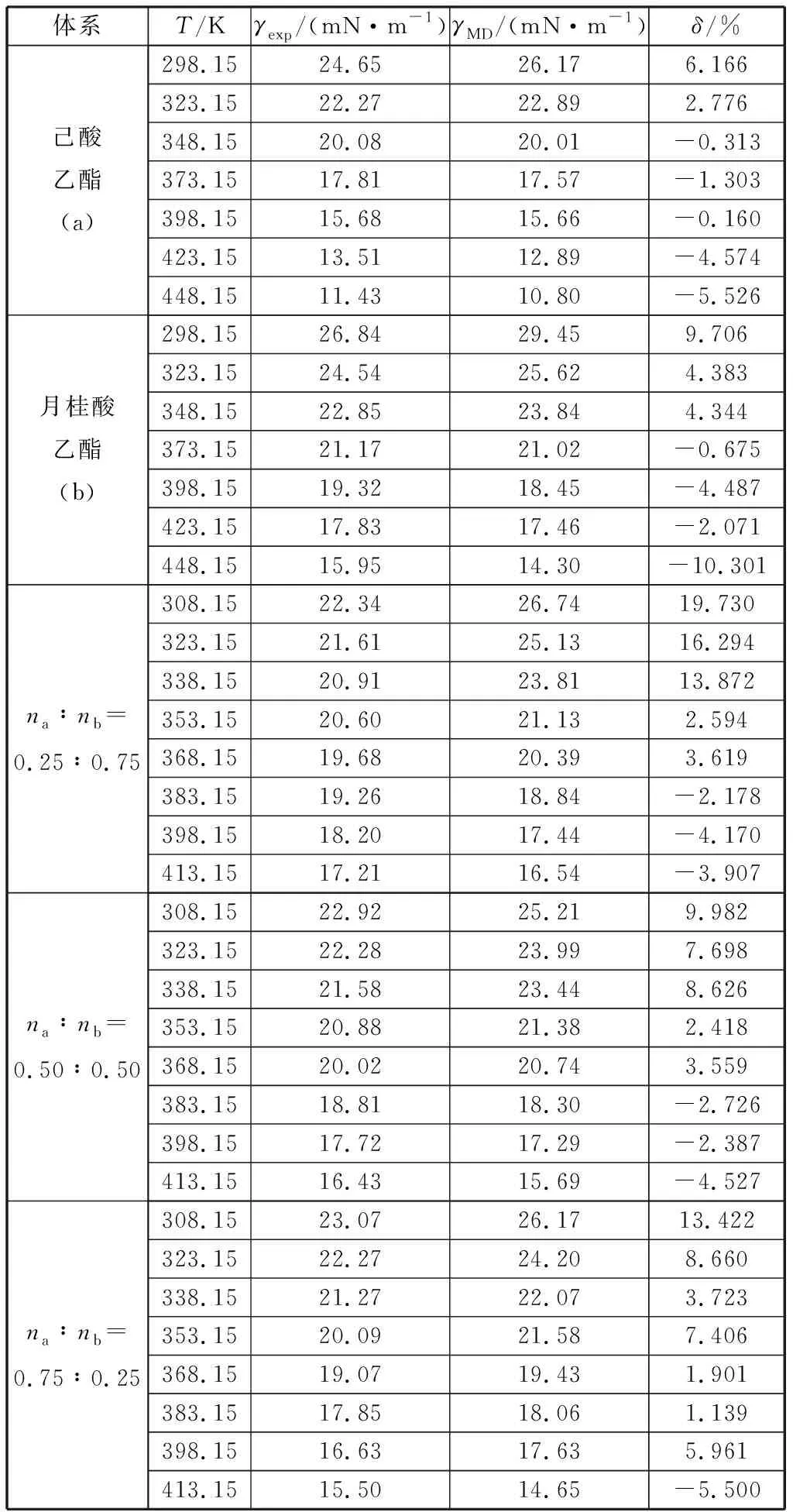
表2 己酸乙酯、月桂酸乙酯及其二元混合物表面张力的实验值γexp[11]与模拟值γMD及其相对偏差δTable 2 Experimental value γexp[11], simulated value γMD and their relative deviation δ for surface tension of ethyl hexanoate, ethyl dodecanoate and their binary mixtures
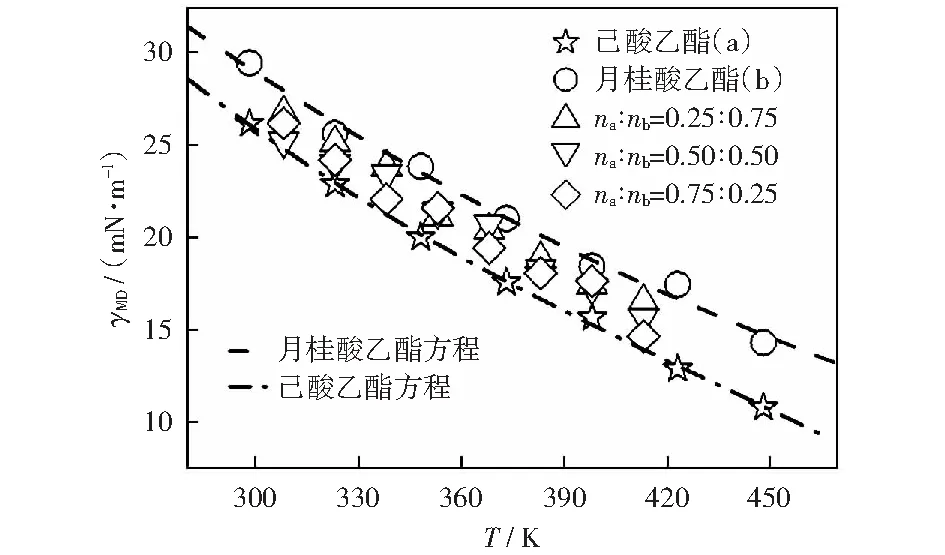
图2 己酸乙酯、月桂酸乙酯在298.15~448.15 K的表面张力模拟数据和拟合方程曲线及其不同配比的二元混合物在308.15~413.15 K的表面张力模拟数据Fig.2 Surface tension simulation data and fitting curves of ethyl hexanoate and ethyl dodecanoate in the temperature range of 298.15~448.15 K and surface tension simulation data of mixtures with different molar fraction ratios in the temperature range of 308.15-413.15 K

图3 己酸乙酯、月桂酸乙酯及其不同配比的二元混合物分别在298.15~448.15 K和308.15~413.15 K温度范围内的表面张力模拟值与实验值的相对偏差Fig.3 Relative deviation of surface tension between simulated value and experimental value for ethyl hexanoate, ethyl dodecanoate and their mixtures with different molar fraction ratios in the temperature ranges of 298.15-448.15 K and 308.15-413.15 K, respectively
2.2 界面性质分析
界面性质的研究对于喷射系统的设计和工艺的优化至关重要。密度的模拟值与实验值的相对偏差较小,最大偏差仅为2.4%,平均偏差为1.4%.在己酸乙酯与月桂酸乙酯摩尔分数比为0.25∶0.75的混合体系中选择6个温度点对其密度分布进行分析,混合物密度在Z轴方向变化情况如图4所示,密度连续且呈对称分布,两侧密度较小的区域为气相区,中间密度均匀区域为液相区,二者的过渡区域为界面区。采用“10-90”的方法计算界面层厚度,即液体密度的10%~90%之间的距离为界面层厚度[12]。图4中D表示界面层厚度,计算结果如表3所示,D(1)、D(2)、D(3)分别代表己酸乙酯与月桂酸乙酯摩尔分数比为0.25∶0.75、0.50∶0.50、0.75∶0.25时界面层的厚度。当体系温度升高时,液相中的分子不断蒸发造成液相区的密度逐渐减小,气相密度逐渐增加,界面层的厚度随温度的升高也逐渐增大;在高温情况下,界面层厚度增加的幅度与己酸乙酯摩尔分数成呈正相关。

图4 不同温度下己酸乙酯与月桂酸乙酯摩尔分数比为0.25∶0.75的混合体系的密度分布Fig.4 Density distribution for a mixed system of ethyl hexanoate and ethyl dodecanoate with a molar fraction ratio of 0.25∶0.75 at different temperatures

图5 不同温度下己酸乙酯与月桂酸乙酯摩尔分数比为0.25∶0.75的混合体系中两种物质的密度分布Fig.5 Density distribution of two substances in a mixed system of ethyl hexanoate and ethyl dodecanoate with a molar fraction ratio of 0.25∶0.75 at different temperatures
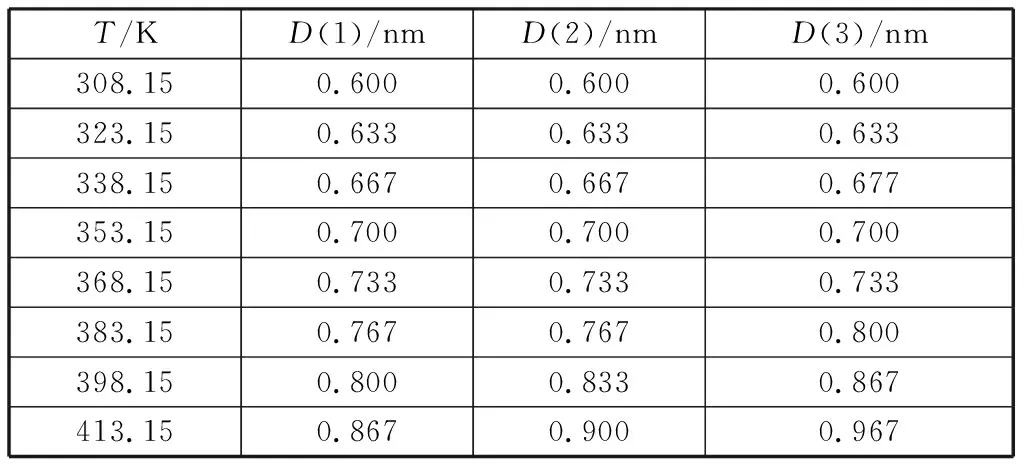
表3 不同温度下体系的界面层厚度Table 3 Interfacial layer thickness of the systems at different temperatures
3 结论
1) 用分子动力学模拟的方法研究了己酸乙酯、月桂酸乙酯及其二元混合物分别在298.15~448.15 K和308.15~413.15 K温度区间内的表面张力,除高温和低温个别温度点与实验数据存在较大的偏差外,其余数据点的偏差均在±10.0%以内。使用方程拟合得到的数据与实验比较的平均偏差为7.8%,在允许偏差范围内,并且方程具有预测性和外延性。
2) 模拟获取了体系的界面性质。随着温度的升高,气-液界面层厚度呈增大趋势,并且在高温条件下,界面层厚度增加量与己酸乙酯摩尔分数呈正相关。分析发现己酸乙酯与月桂酸乙酯混合时,低温条件下己酸乙酯分子倾向于出现在界面区域,随着温度升高现象逐渐减弱。
