马兜铃酸I和马兜铃酸II密度泛函理论研究
2020-06-01焉炳飞祁有国宗轲宁李文佐
焉炳飞,祁有国,宗轲宁,李文佐
(烟台大学化学化工学院,山东 烟台264005)
与西药相比,中草药通常被人们认为是副作用比较小,但自20世纪末国际上报道了含马兜铃酸中草药可能引起肾脏毒性后,对含有马兜铃酸中草药的安全性在国内外引起了广泛关注和讨论[1-5].2017年,有学者在Science子刊《Science Translational Medicine》上发表了一篇封面文章“Aristolochic acides and their derivatives are widely implicated in liver cancers in Taiwan and throughout Asia”,文章指出马兜铃酸及其衍生物对肝癌的发生具有促进作用,该文章的发表引起了巨大的轰动,马兜铃酸迅速成为舆论“焦点”,马兜铃酸的慢性危害更是一个敏感和引起争议的问题[6-7].
马兜铃酸 (aristolochic acid,简称AA) 是一类硝基菲羧酸,天然存在于马兜铃属 (Aristolochia) 和细辛属 (Asarum) 等马兜铃科植物中,其中含量较多的是AAI (8-methoxy-6-nitro-phenanthro-(3,4-d)-1,3-dioxolo-5-carboxylic acid)和AAII (6-nitro-phenanthro-(3,4-d)-1,3-dioxolo-5-carboxylic acid).虽然AAI和AAII有治疗胃痛、缓解高血压、提高白细胞、缓解风湿、治疗水肿、镇痛以及利尿等重要作用,但是其生物毒性却不容忽视,如:诱导基因突变、致癌及肾毒性等[8-9].研究表明,菲环上的硝基 (—NO2) 是AA是主要的毒性基团,其中,AAI中甲氧基 (—OCH3) 对其毒性起着重要的重用[10-11].因此,对AAI和AAII在分子层面上进行细致的研究,对推动中医药事业的传承与现代化发展起着重要的作用.
密度泛函理论是研究分子结构和性质的有效方法[12-15].本工作采用密度泛函理论 (DFT) 的B3LYP、BP86及M06-2X方法,在6-311++G (d,p)基组水平上分别对AAI和AAII分子进行全参数优化,得到了最稳定构型.并在B3LYP/6-311++G (d,p)计算水平上进行了电荷分析及前线分子轨道能级计算.此外,化合物的电离能、亲合能、化学硬度、化学软度、化学势、电负性和亲电性等性质,可能与其稳定性和毒性密切相关,因此本工作对AAI和AAII的这些性质进行了计算.
1 计算方法
通过GaussView5.0软件构建AAI和AAII分子的几何构型.采用DFT的B3LYP[16]、BP86[17]及M06-2X[18]方法,在6-311++G (d,p)基组水平上分别对AAI和AAII分子进行全参数优化,得到了最稳定构型,并在优化构型的基础上用相同的方法进行振动频率分析以确定驻点性质.在B3LYP/6-311++G (d,p)计算水平上计算得到了分子的电荷分布和前线分子轨道 (FMO).
在概念密度泛函 (DFRT) 框架中[19-20],全局性反应活性指数如化学势 (μ) 和化学硬度 (η) 可分别表示为
μ=-χ=(∂E/∂N)ν,
η=(∂2E/∂N2)ν=(∂μ/∂N)ν.
式中,E为体系的总能量,N是体系的电子总数,ν为外部势能,μ可定义为电负性χ的负值.
根据Mulliken原理,当考虑到体系得失电子时的能量变化,运用有限差分近似,化学势μ和化学硬度η可表示为
μ=-(I+A)/2,
η=I-A.
其中,I为第一电离势,A为电子亲和势.又根据Koopman的闭壳层理论,有
I≈-EHOMO,
A≈-ELUMO.
其中,EHOMO和ELUMO分别为最高占据轨道能量和最低空轨道能量.
近来,PARR和LIU等[21]又提出了亲电性指数ω、亲核力差值指数ΔEn、亲电力差值指数ΔEe,分别为
ω=μ2/2η,
ΔEn=-A+ω=(μ+η)2/2η,
ΔEe=I+ω=(μ-η)2/2.
所有计算均采用Gaussian09程序[22]完成.
2 结果与讨论
2.1 分子几何构型
采用B3LYP、BP86和M06-2X方法,在6-311++G (d,p)基组水平上优化得到的AAI与AAII几何构型如图1所示,主要结构参数列于表1,X射线衍射得到的结构参数[23]也列于表1中.3种方法优化得到的AAI和AAII几何构型基本一致,均属于C1点群.菲环体系与五元环结构基本在同一个平面上,由于空间位阻效应使得AAI和AAII的硝基和羧基分别位于这个平面的上方和下方.
从表1可以看出,虽然3种方法计算得到AAI分子的键长与实验值稍有偏差,但是除了C18—O19和C18—O20键长相差较大以外,优化得到的结构与实验结构相差不大.推测C18—O19和C18—O20键长相差较大是因为羟基与其他分子形成分子间氢键.在B3LYP/6-311++G (d,p)计算水平上得到AAI几何结构与实验值相差基本不超过0.000 9 nm,其中,C1—C2、C7—C8、C18—O19、C18—O20、C15—C16、C12—C16键长分别相差0.001 6、0.001 2、-0.002 2、0.004 7、0.001 7、0.001 2 nm;在BP86/6-311++G (d,p)计算水平上得到AAI几何结构与实验值相差较大,绝大多数键长相差超过0.0 10 nm;在M06-2X/6-311++G (d,p)计算水平上得到AAI几何结构与实验值相差基本不超过0.000 9 nm,其中,C1—C2、C3—C4、C1—O7、C18—O19、C18—O20、N21—O22、N21—O23、C15—C16键长分别相差0.001 4、-0.001 2、-0.001 4、-0.002 9、0.003 5、-0.001 3、-0.001 2、0.001 6 nm.此外,考察了3种方法计算得到的结构参数与实验值的线性关系,其相关系数R2分别为0.991 5、0.987 0、0.986 3.考虑到B3LYP方法计算得到AAI几何参数与实验值差别较小,且相关性较好,因此,选择B3LYP/6-311++G (d,p)方法来进行分子性质的计算.此外,从表1可看出,3种方法计算得到的AAII结构参数与AAI相差不大.由于没有找到相关实验值进行比较,但考虑到AAI分子在B3LYP/6-311++G (d,p)水平上计算得到的几何参数与实验值差别较小,且相关性较好,因此,推测B3LYP方法计算得到的AAII几何参数与实验值较为吻合,在进行分子性质计算时也采用B3LYP/6-311++G (d,p)方法.
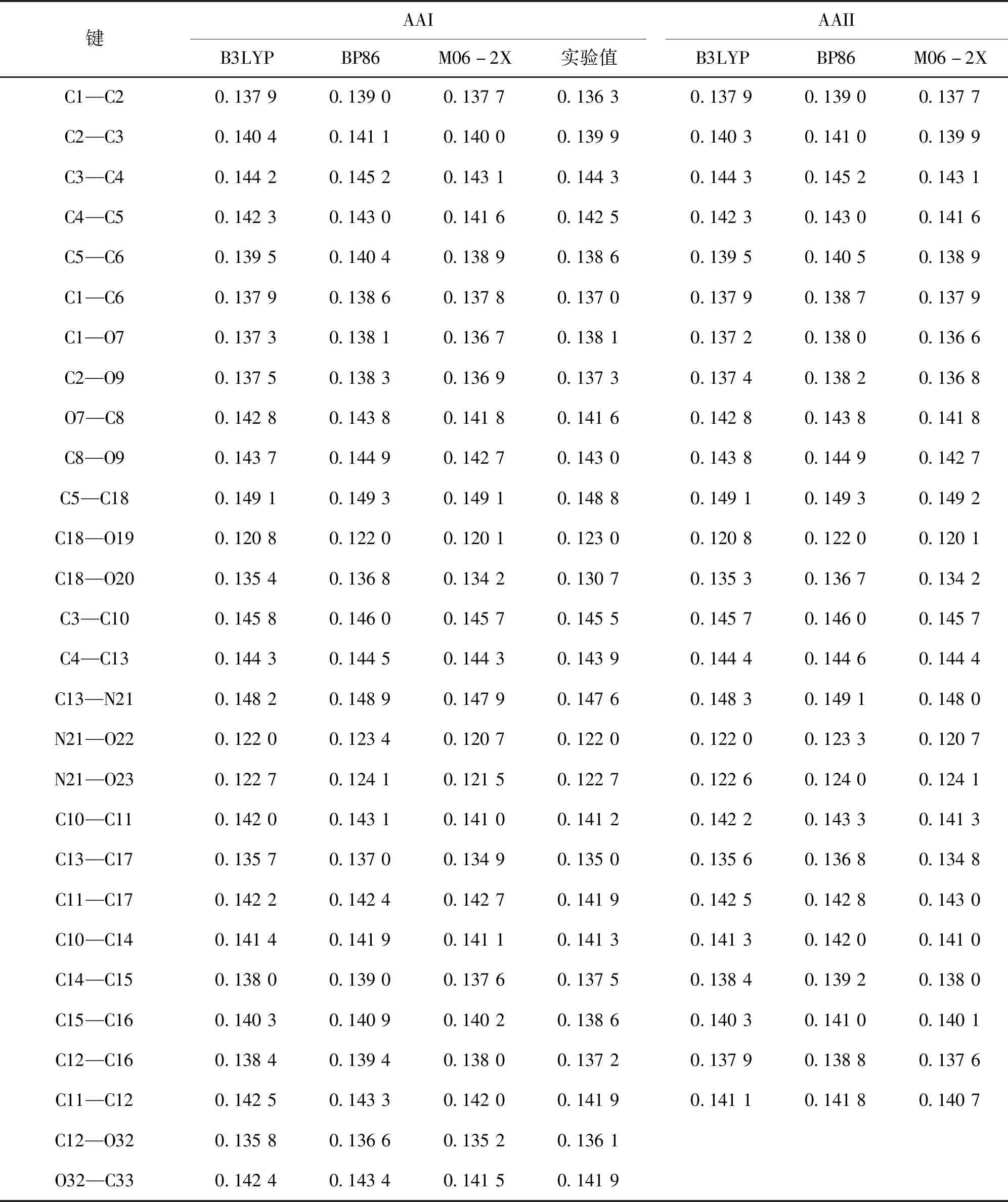
表1 在B3LYP、BP86和M06-2X方法下,6-311++G (d,p)基组水平上计算出AAI与AAII的部分键长参数Tab.1 The bond length of AAI and AAII obtained by B3LYP,BP86 and M06-2X methods with 6-311++G (d,p) basis set nm
2.2 分子电荷分布
药理学研究表明,当受体受到一种药物分子的作用时,必须要通过两者之间电子的相互作用来发挥药效,分析药物分子内的电荷分布可以揭示其与受体的作用点[12].为了比较AAI和AAII毒性的强弱,使用自带的NBO程序对AAI和AAII进行了电荷扫描,其电荷分布情况列于表2.
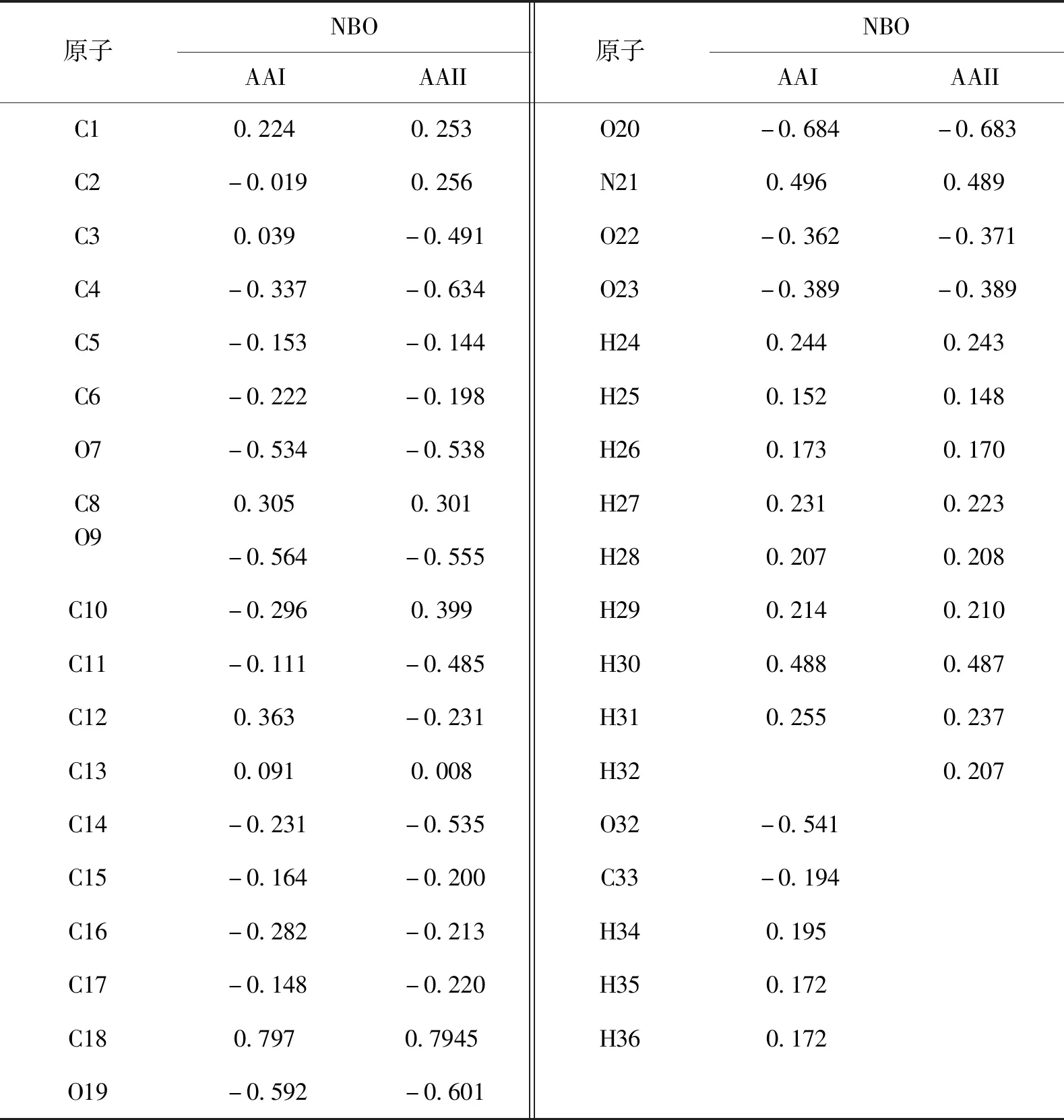
表2 AAI和AAII的电荷分布Tab.2 Charge populations of AAI e
2.3 前线分子轨道特征
1952年日本化学家福井谦一提出前线分子轨道理论[24],能量最高的电子占有轨道(HOMO) 和能量最低的电子未占轨道(LUMO),统称为前线分子轨道,从HOMO可以看出一个分子捐赠电子的能力,而从LUMO可以看到分子获得电子的能力[12].计算得到AAI和AAII的最高占据轨道和最低空轨道的分布如图2所示.根据前线分子轨道理论,分子中电子跃迁最先于HOMO→LUMO,能隙差ΔE越小,电子跃迁的能级越低越容易跃迁,分子活性越高,其中ΔE=ELUMO-EHOMO.
计算结果表明,在B3LYP/6-311++G (d,p)水平得到AAI分子共有88个占据轨道,第88个占据轨道即为HOMO,第89个轨道即为LUMO,其轨道能量分别为-6.109 eV和-2.548 eV,能隙差为3.562 eV;AAII 分子共有80个占据轨道,第80个占据轨道即为HOMO,第81个轨道即为LUMO,其轨道能量分别为-6.303 eV和-2.673 eV,能隙差为3.630 eV.从轨道能量看,AAI和AAII的HOMO和LUMO的对到能量均为负值,说明AAI和AAII的电子状态是稳定的[25].AAI分子的能隙差ΔE小于AAII分子的ΔE,说明AAI比AAII活性高,使得AAI的毒性更大一些.从图2中可看出AAI和AAII前线分子轨道相差不大,HOMO主要分布在菲环及硝基上,尤其在硝基上电子云相对较密集,表现较强的成键性;LUMO则主要分布在菲环及五元环上.
2.4 概念密度泛函活性指数分析
近年来,概念密度泛函活性指数 (如化学势μ、化学硬度η、亲电性指数ω、亲核力差值指数ΔEn、亲电力差值指数ΔEe等)被广泛应用于化合物活性研究.化学势μ越低,化学硬度η越大,分子越稳定,相反分子处于非稳定状态,当化学势μ最高,化学硬度η最小时,分子处于过渡态.亲电指数ω用来表征分子对亲电试剂结合程度,亲核力差值指数ΔEn用来表征分子的亲核活性的大小,亲电力差值指数ΔEe则用来表征分子的亲电活性大小[19-20].上述概念密度泛函活性指数均列于表3中.从表3可看出,AAII的化学势μ较低,化学硬度η较大,即AAII的稳定性比AAI好;AAII的亲电指数ω比AAI要大,即AAII对亲电试剂结合程度比AAI大;AAII的亲核力差值指数ΔEn和亲电力差值指数ΔEe则均比AAI大,即AAII的亲核与亲电的活泼性能要比AAI大.

表3 AAI和AAII的化学势μ、化学硬度η、亲电性指数ω、亲核力差值指数ΔEn、亲电力差值指数ΔEeTab.4 The chemical potential(μ),chemical hardness(η),electro nphilicity (ω),nucleofugality (ΔEn),and electrofugali-ty (ΔEe) of AAI and AAII
3 结 论
本研究通过使用B3LYP、BP86以及M06-2X方法,在6-311++G (d,p)基组水平上对马兜铃酸AAI和AAII分子几何构型优化.与实验值对比发现,B3LYP方法计算得到AAI几何参数与实验值差别较小,且相关性较好.在优化好构型的基础上,用B3LYP/6-311++G (d,p)计算方法进行了电荷分析,计算结果表明,甲氧基的引入使得AAI的硝基N原子正电荷增加,使得AAI的毒性加强;前线分子轨道分析表明,AAI和AAII前线分子轨道相差不大,AAI分子的能隙差ΔE小于AAII分子的ΔE,说明AAI比AAII活性高;此外,采用概念密度泛函活性指数进行了深入分析.本工作为进一步开展马兜铃致病机制的实验研究提供了有用信息,为进一步推动中医药事业的传承与现代化发展打下基础.
