PcG蛋白与血液肿瘤及靶向治疗的相关性研究进展
2020-05-22卞育婕初雅婧袁卫平
卞育婕,初雅婧,袁卫平
(中国医学科学院 北京协和医学院血液病医院(血液学研究所)实验血液学国家重点实验室,天津 300020)
表观遗传修饰由Waddington[1]于1942年提出,现在主要是指不改变核苷酸序列,但基因表达却发生了可遗传、可逆的改变,并最终导致表型改变,主要体现为5种形式:DNA修饰、组蛋白修饰、非编码RNA调控、染色体重塑、核小体定位[2]。PcG蛋白是组蛋白修饰物,在基因转录过程中可以通过压缩染色质结构使转录因子无法接近靶基因启动子或抑制 RNA聚合酶Ⅱ与靶基因结合等方式来抑制转录的起始,在调节细胞增殖、分化、周期等生理进程中发挥重要作用。造血干细胞(hematopoietic stem cell,HSC)的自我更新与分化也与PcG蛋白的调节密切相关[3]。研究表明,白血病干细胞(leukemia stem cell,LSC)起源于HSC[4],PcG蛋白异常表达导致HSC向LSC转变,可能是导致多种血液肿瘤发生的机制之一[5]。因此,PcG蛋白已成为血液肿瘤发病机制和治疗的研究热点,以PcG为靶点的多种药物也已进入临床试验或应用于血液肿瘤的治疗。现就PcG蛋白的组成及分子生物学功能,PcG蛋白异常表达导致的HSC缺陷及血液肿瘤的发生,以及针对PcG蛋白的血液肿瘤的靶点治疗进行综述。
1 PcG蛋白与功能
PcG蛋白最早因调节果蝇体内调控果蝇发育的同源基因(homeobox genes,HOX)而被发现,随后在哺乳动物中也发现存在对应的PcG蛋白同源类似物[6]。不同PcG蛋白组成多梳抑制复合物(polycomb repressive complex,PRC),PRC主要分为PRC1、PRC2、多梳抑制性泛素化酶,见图1。
PRC1又分为经典PRC1和非经典PRC1。经典PRC1核心亚基主要包括环指蛋白1A/B基因(ring finger protein 1A/B,RING1A/B)、CBX(Chromobox)1、PCGF(polycomb group ring finger protein),与其他PCGF亚基相比, B细胞特异的莫罗尼白血病插入位点1(B cell-specific murine leukemia virus insertion site-1,BMI1)基因、黑色素瘤核蛋白-18(melanoma protein 18,MEL-18)基因等与表观遗传修饰关系最为密切。RING1A/B调控组蛋白E3连接酶活性,催化组蛋白H2A的第119位Lys泛素化;CBX连接PRC2主要的催化产物——组蛋白H3第27位赖氨酸上三甲基化(H3K27me3),将PRC1募集至靶基因;PCGF与RING1A/B组成的异二聚体是支撑PRC1装配的主要结构。非经典PRC1不含CBX,而具有与CBX功能类似但不依赖H3K27me3的RYBP(PING1 and YY1 binding protein),与RING1A/B类似具备催化组蛋白H2A第119位赖氨酸单泛素化酶活性赖氨酸去甲基化酶2B[7]。
PRC2是S-腺苷-L-甲硫氨酸依赖性组蛋白甲基转移酶,核心亚基主要包括Zeste基因增强子同源物(enhancer of Zeste homolog,EZH)、胚胎外胚层发育蛋白(embryonic ectoderm development,EED)、Zeste基因抑制子12(suppressor of Zeste 12,SUZ12)、视网膜母细胞瘤结合蛋白46/48(retinoblastoma binding proteins 46/48,RBAP46/48)。EZH2、EED、SUZ12、RBAP46/48的C端共同组成甲基转移酶催化结构域,催化组蛋白H3第27位赖氨酸甲基化生成H3K27me1、H3K27me2、H3K27me3;EED通过与Phe97、Trp364、Tyr365组成芳香结构并与H3K27me3相互作用激活PRC2的组蛋白甲基转移酶活性,从而传播抑制性组蛋白标记;RBAP46/48则可增强甲基转移酶催化活性[5]。
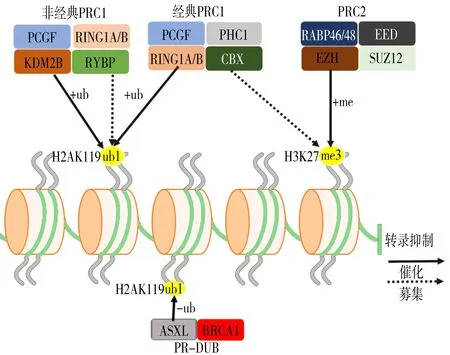
PRC:多梳抑制复合物;PCGF:多梳基因环指蛋白基因;RING1A/B:环指蛋白1A/B基因;PHC1:多同源同系物1;RBAP46/48:视网膜母细胞瘤结合蛋白46/48;EED:胚胎外胚层发育蛋白;KDM2B:赖氨酸去甲基化酶2B;RYBP:PING1 and YY1 binding protein;CBX:Chromobox;EZH:Zeste基因增强子同源物;SUZ12:Zeste基因抑制子12;H2AK119ub1:组蛋白H2A第119位赖氨酸进行单泛素化蛋白;H3K27me3:组蛋白H3第27位赖氨酸上三甲基化;ASXL:Additional sex comb;PR-DUB:多梳抑制性泛素化酶
图1 PcG蛋白与组蛋白相互作用示意图
多梳抑制性泛素化酶是一种组蛋白去泛素化酶,可去除组蛋白H2A第119位赖氨酸进行单泛素化蛋白(H2AK119ub1)的单泛素化标记,多梳抑制性泛素化酶的缺失导致体内H2AK119ub1水平增加近10倍并导致果蝇中HOX基因的去阻遏。在果蝇中,Calypso与ASXL(additional sex comb)组成配偶体,Calypso是分布在蛋白C端水解酶类的去泛素化酶,需要ASX的存在才具备酶活性;在哺乳动物中,BRCA1相关蛋白是Calypso同源物,与ASX同源物1-3结合成配偶体,同样BAP1也需要在ASX同源物蛋白存在的情况下才具备酶活性[8]。
PRC2定位至染色体后催化组蛋白产生H3K27me3,PRC1亚基CBX连接到H3K27me3后催化组蛋白产生H2AK119ub1,H2AK119ub1可增强JARID2(Jumonji and AT-rich interaction domain containing 2)活性募集更多PRC2至染色体,PRC2继续募集PRC1,进而形成放大的正反馈信号环路;PRC1催化产物H2AK119ub1通过与RNA聚合酶Ⅱ结合来抑制靶基因转录,进而阻断RNA聚合酶Ⅱ的置换,抑制靶基因转录;PRC2催化产物H3K27me3压缩染色体来抑制靶基因转录;多梳抑制性泛素化酶使H2AK119ub1去泛素化来抑制PRC1部分功能,三者共同作用调节靶基因的表达。
2 PcG蛋白与造血
造血稳态是HSC自我更新和分化动态平衡的结果,PcG蛋白通过调节HSC自我更新与分化维持造血稳态。遗传小鼠模型和患者突变的研究强调了染色质调节在正常和恶性血细胞生成中的关键影响,因此,PcG蛋白异常表达可引起HSC数量和功能异常,无法维持机体正常造血,进而导致多种血液肿瘤的发生。
2.1PRC1与HSC 敲除RING1A/B,在体外导致Lin-细胞集落出现生长缺陷[9];在体内,小鼠骨髓中HSC耗竭、脾脏中不同程度HSC或髓系祖细胞过度增殖,并发生再生障碍性贫血。PcG蛋白被认为主要通过转录抑制来调节基因表达,但有研究表明,在细胞有丝分裂过程中,RING1A/B可调节与启动子结合的染色质相关蛋白的泛素化,当细胞进入G1期,这种修饰对于标记基因的表达是必需的;敲除RING1A/B,其所标记的转录相关基因无法表达,细胞周期阻滞在G1期,HSC无法自我更新与分化[10]。
在小鼠细胞体外培养时,CBX7过表达可提高HSC、多潜能祖细胞的自我更新能力,但CBX7过表达抑制致死剂量照射小鼠体内HSC造血重建,CBX7缺陷导致HSC的增殖和集落形成能力下降,提示CBX7可维持HSC、多潜能祖细胞的自我更新能力;CBX2过表达对HSC增殖无影响,CBX4或CBX8过表达导致HSC趋向分化和耗竭[11]。在利用CBX过表达细胞构建的HSC竞争移植模型中,CBX7过表达HSC具备显著的竞争性再生优势;而CBX2过表达小鼠体内仅完成B淋巴细胞重建,提示CBX2可促进淋巴细胞生成,而非髓系细胞生成;CBX4或CBX8过表达HSC均未能促成长期造血重建[12]。总之,不同CBX蛋白的存在赋予PRC1不同的靶基因选择性,共同维持HSC的自我更新和分化之间的动态平衡。
BMI1-/-小鼠胎肝HSC数量正常可以维持正常造血,但出生后骨髓HSC减少、全血细胞减少并在2个月内死亡;将BMI1-/-小鼠胎肝HSC移植至致死剂量照射小鼠体内,肝脏无法维持长期造血重建[13];BMI1过表达,促进HSC体外造血分化中红系和混合系集落的产生,提示BMI1在维持HSC自我更新和分化中发挥重要作用[14]。BMI1缺陷的HSC中Ink4a-Arf上调,其编码两种蛋白质p16Ink4a和p19Arf。p16Ink4a作为细胞周期蛋白依赖性激酶抑制剂起作用,通过激活Rb途径阻碍细胞周期进程,而p19Arf协调p21/p53介导的细胞周期停滞和凋亡。BMI1-/-HSC由于p16Ink4a和p19Arf去阻遏导致HSC无法自我更新与分化,提示抑制Ink4a-Arf是BMI1调控HSC自我更新与分化的有效机制;BMI1通过在氧化应激时保护DNA免受损伤来调控HSC自我更新与维持的功能,在BMI1缺陷的情况下,HSC中活性氧类水平升高,导致细胞凋亡增加,HSC数量减少和活性降低[15]。
MEL-18和BMI1同属于PCGF蛋白,两者氨基酸序列高度同源但功能不同。BMI1在未成熟HSC中高表达,随着细胞分化表达减少,而MEL-18随着HSC分化表达增加;虽然BMI1-/-的小鼠胎肝HSC功能缺陷,但MEL-18-/-小鼠胎肝HSC功能不受影响,而B细胞数量减少[16-17]。因此,MEL-18可能主要在更多分化细胞中发挥调节作用而不参与HSC的自我更新与维持。
Rae28-/-小鼠胎肝HSC数量从胚胎期14.5 d逐渐降低,围生期死亡率升高;移植Rae28-/-小鼠HSC至致死剂量照射的小鼠体内,受体小鼠的骨髓造血功能衰竭,表现为HSC及各系细胞减少,无法完成功能性造血重建,提示Rae28在维持胎肝HSC的自我更新与分化方面发挥重要作用,但Rae28对成体HSC功能的影响仍有待阐述[18]。研究表明,Rae28缺陷会抑制泛素-蛋白酶体介导的Geminin降解,Geminin是DNA复制许可因子Cdt1的抑制剂,因此累积的Geminin抑制DNA复制,从而抑制HSC自我更新[19]。
2.2PRC2与HSC Zeste蛋白的两种同源类似物EZH1、EZH2拥有相同SET催化结构域,催化H3第27位Lys甲基化,两者既有相同的作用靶点又有各自选择性作用靶点,EZH1、EZH2双敲除的HSC,H3第27位Lys不能被甲基化,仅敲除EZH2后H3第27位Lys仍能被甲基化,但严重损伤了HSC的自我更新能力,EZH1可以补偿成体HSC中的EZH2缺陷,但不能补偿胎肝HSC中EZH2缺陷。
研究表明,EZH1-/-的小鼠骨髓造血衰竭表现为HSC及各系细胞减少,提示EZH1在维持HSC更新中发挥重要作用[20];EZH1促进胚胎干细胞分化为多种淋巴细胞,并导致体内功能确定性的成熟HSC出现,在哺乳动物早期胚胎阶段抑制胚胎干细胞的多能性[21]。EZH1在成体HSC中特异性表达,并且随HSC分化表达减少,主要调控成体HSC造血,对胎肝HSC的造血调控是非必需的[22]。
EZH2在人和小鼠骨髓和胎肝细胞中广泛表达,为重要的HSC调控因子,在连续HSC移植中HSC逐渐耗竭,但EZH2过表达可维持HSC自我更新,实现连续移植后HSC仍能大量增殖以维持受体鼠体内造血重建[23]。但EZH2缺陷的HSC仍具备较强的移植能力,受体鼠淋巴细胞发育和免疫球蛋白可变区基因片段重排受到影响,淋巴谱系发育缺陷,而髓系发育并未受到影响。研究表明,EZH过表达抑制了HSC中p16Ink4a、p19Arf、促分化的基因(如DNA连接蛋白抑制基因2、亚硫酸氧化酶基因7)、促凋亡基因(如NADH氧化酶基因A、p21)等表达来维持HSC自我更新与分化[24]。
研究表明,EED突变纯合子胎肝HSC的H3K27me3明显减少,小鼠于胚胎期死亡;与突变纯合子相比,EED突变杂合子HSC的克隆形成能力和骨髓再生活性增加,提示EED在维持HSC自我更新中发挥重要作用[5];EED-/-小鼠脾脏体积明显缩小,脾脏和骨髓苍白,血细胞计数呈血白细胞和红细胞减少,骨髓淋巴细胞和髓系细胞均减少,提示EED调节胚胎发育和造血平衡[25]。SUZ12-/-小鼠胎肝HSC和成体HSC均显示造血功能缺陷,体内各系细胞生成减少,提示SUZ12在造血稳态的调节中发挥重要作用[26]。
3 PcG蛋白与血液肿瘤
3.1PRC1与血液肿瘤 血液肿瘤是起源于HSC的恶性增殖性肿瘤,其发生是一个多步骤过程。参与细胞周期调控的基因活性异常是细胞恶性转化的先决条件之一,许多恶性细胞也已失去正常的细胞周期进程。原癌基因和抑癌基因(如Ink4a-Arf)通常在癌细胞中失调,PcG蛋白的异常表达会导致包括原癌基因和抑癌基因在内的靶基因发生突变、缺失和过表达等遗传改变,这些异常的遗传改变驱动抑癌基因的失活或致癌基因的启动,导致HSC无法自我更新与分化来维持机体正常造血,进而出现以异常造血为主要表征的血液肿瘤的发生。但PcG蛋白功能复杂,在不同血液肿瘤中以不同分子机制分别发挥致癌或抑癌作用。
RING1A/B通常在骨髓增生异常综合征(myelodysplastic syndromes,MDS)、急性髓系白血病(acute myeloid leukemia,AML)、非霍奇金淋巴瘤、弥漫大B淋巴瘤等血液肿瘤中过表达并且与预后不良相关。研究发现,幼年型髓单核细胞白血病患者的肿瘤细胞系中存在RING突变体-CBL-C384R,其上调细胞内Janus激酶2和LYN激酶表达,并增强粒细胞-巨噬细胞集落刺激因子的下游信号转导,是幼年型粒单细胞白血病发病的可能机制之一[23]。
CBX7过表达与C-myc共同作用导致T细胞或B细胞淋巴瘤[27];敲除CBX8或抑制其与点突变的结合可以干扰HOXA9基表达上调从而阻止MLL-AF9白血病的发生;而CBX2与CBX4与白血病的发展无关[28]。
BMI1的过表达常见于MDS、AML、慢性粒细胞白血病和各种类型的淋巴瘤患者[24,29]。BMI1可能协同诱导白血病转化,Sall4是一种导致AML发生的致癌基因,与BMI1启动子结合并诱导其表达;HoxA9-Meis1白血病小鼠模型中,HOXA9和Meis1致癌基因可诱导小鼠骨髓HSC转化为LSC。Hoxa9-Meis1转导的BMI1-/-胎肝HSC移植入致死剂量照射小鼠,仅能在初次移植受体体内诱导AML,连续移植后无法诱导受体鼠AML的产生,提示BMI1对维持LSC至关重要[30-31]。BMI1通过抑制p16Ink4a、p19Arf维持LSC的恶性增殖以及导致恶性肿瘤的发生。
有研究通过评估多例AML患者中多个PcG家族成员的表达,发现BMI1过表达广泛存在,而Mel-18是少数几个未显示过表达的PcG基因之一[32]。此外,在多例血液恶性肿瘤患者HSC中存在Rae28变异,而且Rae28的失活与肿瘤恶性程度有关并提示不良预后[19]。
3.2PRC2与血液肿瘤 EZH2的催化产物H3K27me3抑制许多靶基因转录,其功能障碍被认为可能作用于原癌或抑癌基因发挥致癌或抑癌作用。EZH2突变的研究表明,在多种肿瘤中观察到EZH2基因的体细胞突变[33]。在淋巴瘤中存在EZH2的酪氨酸641(Y641F)的杂合点突变,并且已经在淋巴瘤中报道了EZH2过表达或EZH2的SET结构域中的突变,导致H3K27me3增加[34];滤泡性淋巴瘤EZH2的功能获得性突变导致H3K27me3增加从而抑制周期依赖性激酶基因1A、T细胞/淋巴瘤基因1A等基因表达[35]。相反,在MDS、骨髓增殖性肿瘤、急性T淋巴细胞白血病等疾病中存在EZH2的缺失、移码、无义和错义突变,提示EZH2的功能缺失性突变与肿瘤的发生发展有关,EZH2可能发挥肿瘤抑制作用[36];与在患者中的这些发现一致,在EZH2缺陷的小鼠模型中,研究者发现EZH2缺陷上调了RUNX1(RUNX family transcription factor 1)基因的表达,并成功诱导了MDS的发生[8]。
尽管EED和SUZ12突变频率较EZH2低,但也与多种肿瘤形成相关。EED和SUZ12协同调节EZH2的甲基转移酶活性,EED或SUZ12异常将通过影响EZH2酶活性导致肿瘤发生。研究发现,EED单倍体缺陷小鼠自发发生MDS、骨髓增殖性肿瘤[37];EED杂合小鼠较纯合小鼠更易患恶性肿瘤,并与Evi1过表达共同导致白血病发生[38]。SUZ12突变存在于多种肿瘤中,尤其是套细胞淋巴瘤(mantle cell lymphoma,MCL)存在大量的SUZ12突变体的基因扩增产物;SUZ12突变抑制Ink4,促进MCL细胞增殖和抑制细胞凋亡[26]。
4 针对异常PcG蛋白功能的靶点治疗
PcG在正常造血过程中发挥重要作用,异常表达的PcG亚基导致造血系统异常,从而导致多种血液肿瘤的发生。因此,靶向异常的PcG亚基并给予纠正是血液肿瘤靶向治疗的重要方向,靶向异常PcG亚基的相关药物及临床试验总结见表1。

表1 针对PcG蛋白的靶向治疗概述
SAH-EZH2:稳定的EZH2α-螺旋肽;CBX:Chromobox;RING:环指蛋白;EZH:Zeste基因增强子同源物;BMI1:B细胞特异的莫罗尼白血病插入位点1;EED:胚胎外胚层发育蛋白;CML:慢性髓系白血病;MDS:骨髓增生异常综合征;AML:急性髓系白血病;DLBCL:弥漫大B细胞淋巴瘤;MCL:套细胞淋巴瘤;FL:滤泡性淋巴瘤;NHL:非霍奇金淋巴瘤;MM:多发性骨髓瘤;MLL:混合谱系白血病;-:已临床应用
4.1针对PRC1的靶点治疗 应用伊马替尼治疗CML,患者肿瘤细胞中CBX6、CBX7和BMI1均升高,但作用不同。CBX6、7发挥抑癌基因作用,诱导致癌基因沉默和肿瘤细胞死亡,因此大部分患者对伊马替尼治疗有短期的良好反应[39]。但有部分肿瘤细胞发生逃逸并在BMI1作用下自我更新促进肿瘤进展,即预示耐药性的出现及预后不良[40]。组蛋白乙酰化酶抑制剂伏立诺他通过驱动小泛素化修饰蛋白来介导CBX2的降解,导致CBX缺陷的白血病细胞增殖受损,以达到延缓和抑制白血病进展的目的[41]。地西他滨治疗可显著降低MDS和AML患者肿瘤细胞内RING1A/B和EZH2的表达水平,有效延长生存期[30]。
PTC-208、PTC-029、PTC596是近年来开发的靶向BMI1的新型小分子选择性抑制剂,具有不同的作用模式[42]。PTC-028诱导BMI1的磷酸化,加速BMI1降解;PTC-209干扰BMI1的转录后调控从而下调BMI1;MCL-1是AML中关键的抗凋亡Bcl-2家族蛋白,在AML细胞中特别是在CD34+CD38-干细胞、祖细胞群高表达,BMI1通过磷脂酰肌醇-3-激酶/蛋白激酶B上调MCL-1,而PTC596通过磷脂酰肌醇-3-激酶/蛋白激酶B通路抑制MCL-1,促进AML干细胞、祖细胞凋亡来治疗AML。PTC-209由于其有限的效力和较差的药动学特性尚未进入临床试验。PTC596已在晚期实体瘤患者(NCT02404480)中完成了Ⅰ期临床试验[43]。PTC596可有效杀死AML患者来源的干细胞、祖细胞,显示出良好的安全性。
4.2针对PRC2的靶点治疗 EZH2广泛调节基因的表达和调控细胞功能,在不同肿瘤中发挥致癌基因或抑癌基因的功能。因此,EZH2是肿瘤的潜在治疗靶点,目前针对EZH2致癌作用的EZH2抑制剂的临床前和临床试验正在进行,且EZH2抑制剂的有效性在淋巴瘤和多发性骨髓瘤中也得到证实。
3-去氮腺苷的环戊醇类似物竞争性抑制S-腺苷同型半胱氨酸水解酶活性而导致S-腺苷同型半胱氨酸在细胞内堆积,S-腺苷同型半胱氨酸与EZH2的酶活性中心结合而抑制EZH2的甲基转移酶活性,解除EZH2对抑癌基因的抑制,在急性早幼粒细胞白血病、淋巴瘤中发挥抑癌作用,但有研究指出3-去氮腺苷的环戊醇类似物应用于动物模型时存在毒性作用[42]。GSK126[44]和EPZ005687[45]也是选择性S-腺苷甲硫氨酸竞争性小分子抑制剂,可以结合野生型EZH2和Y641,导致H3K27me3表达上调和被EZH2抑制的靶基因表达上调,在体外细胞培养和动物模型中有效抑制弥漫大B淋巴瘤的进展。EZH2抑制剂EPZ6438(tazemetostat)可降低H3K27me3的水平,从而抑制EZH2突变导致的非霍奇金淋巴瘤生长。EPZ6438目前正应用于评估B细胞淋巴瘤和晚期实体瘤患者的Ⅰ/Ⅱ期临床试验(NCT01897571)[46]。EPZ011989是一种具有有效药动学特性的口服的选择性EZH2抑制剂。EPZ011989在人B细胞淋巴瘤的小鼠异种移植模型中显示出显著的肿瘤生长抑制作用[47]。EBI-2511是一种基于苯并呋喃衍生的EZH2抑制剂,在小鼠的肿瘤异种移植模型中抑制肿瘤细胞增殖,并且正在临床前开发用于治疗与EZH2突变相关的肿瘤[48]。研究表明,稳定的EZH2 α-螺旋肽(stabilized alpha-helix of EZH2,SAH-EZH2)通过破坏EZH2-EED复合物和降低EZH2蛋白水平选择性地抑制H3K27me3,导致MLL-AF9白血病细胞的生长停滞和分化[49]。另一种抑制EZH2与PRC2复合物相互作用的抑制剂CPI-1205目前正在一项B细胞淋巴瘤患者的Ⅰ期临床试验中进行评估[50],针对EZH2的GSK-2816126[51]也进入各种恶性血液肿瘤的临床试验。
除EZH2特异性抑制剂外,因EZH1部分代偿EZH2功能,靶向EZH1/2可能是一种很有前景的治疗方法。UNC1999靶向抑制EZH1/2使H3K27甲基化减少和H3K27乙酰化增加,导致周期依赖性激酶基因2A等细胞周期调控基因和发育基因的去阻遏,诱导一系列抗白血病作用(如肿瘤细胞的凋亡),从而抑制MLL白血病的发展[52]。OR-S1可选择性抑制EZH1/2的组蛋白甲基转移酶活性,激活Wnt信号通路来减少LSC以抑制白血病的进展[53]。DS-3201b是一种针对EZH1和EZH2的特异性抑制剂,已在临床前研究中证实了对复发或难治性非霍奇金淋巴瘤、人T细胞白血病-淋巴瘤等多种血液恶性肿瘤的抗肿瘤活性[54]。
A-395是一种破坏EED-H3K27me3相互作用的小分子抑制剂,在小鼠淋巴瘤异种移植模型中显示出肿瘤抑制功效,在人骨肉瘤细胞中证实该药物减少H3K27me3积累,主要作为组蛋白甲基转移酶抑制剂起作用[55]。小分子抑制剂阿司咪唑也可抑制EED/EZH2相互作用,破坏PRC2复合物的稳定性,从而减少淋巴瘤细胞的增殖[56]。
5 结 语
来自体外和体内模型的累积证据以及临床研究共同证实PcG蛋白参与多种血液肿瘤的发生和发展。PcG蛋白的缺失或过表达通过抑制抑癌基因和上调致癌基因表达,促进正常HSC向肿瘤干细胞转化,是血液肿瘤发生和治疗后复发的机制之一。因此,多种针对不同PcG蛋白的靶向药物也已经引入血液肿瘤的临床试验,不同于传统肿瘤的治疗靶点的不可及性,在血液肿瘤的治疗中引入靶向PcG蛋白的靶向药物能够更有效和特异性地到达治疗靶点,目前已有针对PcG蛋白亚基的靶点药物进入临床试验或应用于血液肿瘤患者的临床治疗并取得初步疗效。PcG蛋白功能复杂,许多复杂功能仍有待进一步阐述,因此,详细了解造血系统中PcG蛋白的复杂功能将为血液肿瘤治疗提供新的靶点,以及为血液肿瘤靶向药物耐药问题提供新的思路。
