Peutz-Jeghers综合征伴子宫恶性肿瘤家系报道并文献回顾
2020-05-21邢洁杨悦郑爱文黄晶晶
邢洁 杨悦 郑爱文* 黄晶晶
Peutz-Jeghers综合征(Peutz-Jeghers syndrome,PJS)又称为黑斑息肉综合征,是一种较为罕见的常染色体显性遗传病,以胃肠道多发息肉和皮肤、黏膜色素沉着为特征。该疾病最早在1921年和1949年分别由Peutz和Jeghers发现并报道,其发病率为 1/20 万 ~1/12 万[1],50%PJS 患者有家族史[2]。浙江省肿瘤医院最新诊治PJS综合征伴子宫颈粘液腺癌的家系患者,现结合文献复习报道如下。
1 临床资料
例1,患者,女,45岁,已婚,既往体质良好,无吸烟饮酒习惯,无毒物及放射性物质接触史。因“便血1月”入院。查体:口唇、手指和手掌、脚趾可见散在、多发色素沉着(见图A、B)。妇检外阴无殊,阴道黏膜光滑,宫颈光,子宫平位,常大,左侧盆腔及10cm肿块,活动度可。直肠黏膜光,指套无染血。2019年5月查糖类抗原199(CA199)2572.43U/ml。液基薄层细胞检测(TCT):未见上皮内病变或恶性细胞(NILM)。人乳头瘤病毒(HPV):均呈阴性。肠镜示:结肠多发息肉(左半结肠为主),乙状结肠息肉癌变考虑。全腹CT:(1)盆腔内囊性灶,考虑附件来源恶性肿瘤可能;(2)腹、盆腔大量积液,盆底腹膜结节,考虑转移;(3)宫颈多发囊肿。初步诊断:盆腔肿瘤(卵巢恶性肿瘤?),家族性结直肠息肉病,胃息肉病,腹水。2019年5月22日行卵巢癌根治术:子宫全切+双附件切除+大网膜+盆腔淋巴结切除+卷地毯式盆腔腹膜剥除+减瘤术+肠粘连松解术+全结肠切除+回肠直肠端侧吻合+肠修补(小肠直肠)+膈肌肿瘤切除+膈肌修补+双侧输尿管松解。术中探查见:淡黄色粘液样腹水约5000ml,分离肠管粘连,右膈肌表面多发片状结节(7cm×8cm),左侧膈肌表面多发片状结节(7cm×7cm),肝表面1枚结节(1cm)。小网膜1枚结节(3cm),大网膜成饼状,结肠系膜多发结节(0.5~2cm),小肠表面及系膜散在结节(1cm)。左卵巢可见囊实性肿块(10cm),表面光滑,肿块与周围组织无明显粘连。右侧卵巢大小3cm×2cm×2cm表面光。子宫体常大。左右结肠旁沟、左右盆腔腹膜散在结节(1~2cm),膀胱反折腹膜、子宫直肠窝融合成片状增厚脓苔样肿瘤病灶。术后无肉眼残余灶。术后病理:(1)宫颈内生型粘液腺癌,累犯宫体内膜至浅肌层,累犯左卵巢及(右半)结肠浆膜纤维、脂肪组织,转移或浸润至(膀胱反折腹膜、子宫直肠窝、大网膜、右结肠旁沟、小肠系膜、小肠表面、骶前、肝脏表面、右侧横膈、小网膜、左侧横膈)纤维、脂肪组织内及(左盆腔)1/2只、(右盆腔)0/3只、(回肠周)0/4只、(结肠系膜根部)2/5只、(肠系膜)1/3只、(回盲部)1/7只、(结肠上组)0/2只、(结肠中组)0/7只、(结肠下组)0/4只、(直肠系膜)1/1只淋巴结。(2)(右)卵巢组织内见性索间质肿瘤(考虑为环状小管性索肿瘤)。免疫组化:ER(宫体)、PR(宫体)、Vim(宫体 -)、PAX8(宫体少量+):WT-1(宫体)、CK7(宫体+)、P16(宫)P53(宫体+~++约60%)、Ki-67(宫体+,约 70%)、P16(宫颈部分 +)、P53宫颈 +~++约 70%)Ki-67(宫颈+,约80%)、ER(宫颈-)、PR(宫颈)、Vim(宫颈)、CDX-2(宫颈+)、CEA(宫颈+)、CK20(宫颈-)、CK7(宫颈+)(见图C、D、E)。修正诊断:宫颈恶性肿瘤Ⅳ期(粘液腺癌),家族性结直肠息肉病,胃息肉病。拟术后行6~8程辅助化疗。2019年6月18日行第1程TP方案化疗:紫杉醇230mg ivgtt d1+顺铂85mg ivgtt d1,化疗后2周患者出现恶心、呃逆,较剧,难忍,伴呕吐,呕吐物为胃内容物,伴乏力明显。2019年7月8日腹部平片提示肠梗阻。查CA199 50492.16U/ml。CT:(1)卵巢癌根治术后,盆腔积液;(2)肝右叶多枚结节灶,转移灶考虑,腹膜、肠系膜及网膜增厚、模糊,腹腔见大量积液影,转移考虑。右侧心膈角肿大淋巴结。双侧胸腔少量积液。考虑患者原发抗药,予小肠减压及对症治疗后患者肠梗阻未好转,复查CA199>70000U/ml,于2019年8月中旬死亡。将患者宫颈术后石蜡包埋组织切片样本、患者本人(6#)及其两女(10#、11#)共三人血液样本一并送至北京泛生子医学检验所有限公司行基因检测,对患者(6#)组织切片和血液样本分别进行实体瘤825基因检测(含830个基因全外显子区和部分内含子区域内所有点突变、插入缺失突变、拷贝数变异及重排突变)和大片段重复或缺失检测(MLPA技术),对两女儿(10#、11#)血液样本则同时进行139遗传易感基因检测和MLPA技术检测,三人检测结果点突变如表1所示,母女三人血液样本MLPA检测结果显示仅患者本人(6#)STK11基因2、3号外显子缺失,两女儿(10#、11#)STK11基因正常。(见图 H~M,表 1~2)。
例2,患者,女,48岁,已婚。既往体质良好,无吸烟饮酒习惯,无毒物及放射性物质接触史。因“阴道分泌物增多半年余”,于2017年6月就诊妇科检查:外阴无殊,阴道黏膜光滑,内见中等量白色透明分泌物,宫颈肿瘤5cm,子宫前位,常大,盆腔未及肿块,直肠黏膜光,指套无染血。宫颈活检:宫颈粘液腺癌。诊断:宫颈粘液腺癌IB2期。2017年6月5日行宫颈癌根治术。术后病理:宫颈粘液腺癌,浸润宫颈全层,向上侵犯子宫体下段,见脉管内癌栓,双侧宫旁见癌累及,左输卵管上皮间癌累及,盆腔及腹主动脉旁淋巴结均未见转移(0/55);ER/PR(-),Ki67(<1%+)(见图F、G)。术后修正诊断:宫颈粘液腺癌IIA2期。术后予夹心治疗:盆腔外照射联合3疗程化疗(紫杉醇d1+顺铂d1~3)。2018年9月患者复查CA199 680.87U/ml;全腹CT:子宫颈癌术后,阴道残端增厚。诊断宫颈腺癌术后放化疗后复发。行吉西他滨dl、8+顺铂d1-3方案化疗。化疗2程后患者出现右腰部疼痛。行全腹CT:子宫颈癌术后,阴道残端肿瘤较前增大,右侧输尿管扩张伴右肾重度积水。遂行右肾造瘘术。术后患者停止化疗,定期复查。2019年7月26日全腹。将患者(4#)及其两女(8#、9#)共三人唾液样本一并送至北京泛生子医学检验所有限公司同时进行139遗传易感基因检测和MLPA技术检测,点突变检测结果如表1所示,MLPA检测结果显示患者本人(4#)及小女(9#)均STK11 Exon2~3缺失,大女儿(8#)STK11基因正常。(见图H~M,表 1、2)。

图A、B:案例1患者黑色素斑(A:手部;B:唇部);图C、D:案例1患者病理结果(C:宫颈肿物;D:直肠息肉);图E:案例1患者术后直肠息肉;图F、G:案例2患者宫颈肿瘤病理结果

表1 家系基因点突变检测结果

表2 家系STK11基因缺失检测结果
2 讨论
2017年,美国国立综合癌症网络(NCCN)的遗传性家族性结直肠癌高危风险评估指南[3]中,符合以下任意两项即可诊断为PJS:(1)两个或多个PJS型小肠息肉;(2)口腔、嘴唇、鼻、眼、生殖器或手指的皮肤、黏膜色素沉着;(3)PJS家族史。本案例患者为母女三人,母亲已故,生前皮肤黏膜有黑色素斑,死因为子宫恶性肿瘤伴腹水,2例患者为两姐妹,均有黑色素斑,均患宫颈粘液腺癌、胃肠道多发息肉,病例2患者与一正常男子婚配后有一女已表现出黑色素斑,患者弟弟表现肠道息肉且有黑色素斑特征,无子女,家族图谱如图F所示,根据诊断标准可确诊此家系为PJS。
抑 癌 基 因 STK11(Serine threonine kinase 11,MIM#602216)基因突变被认为是PJS的致病原因[4],目前已发现包括错义突变、缺失突变、插入突变200余种STK11基因致病突变[5],但突变类型和恶性肿瘤的关系尚无定论。约50%的PJS患者携带STK11基因突变。基因检测一方面有助于本案对PJS的诊断,尤其是对STK11基因的突变检测,另一方面可分析患者家系成员之间遗传性基因突变情况,为此,对本案例全部家系成员(父母双亡、弟弟失访除外)利用探针杂交捕获技术和Illumina高通量基因检测技术进行830个实体瘤热点突变基因及139个遗传易感基因检测(结果见表1),6位受检者的检测结果各异,PJS患者无论在同代之间(4#和6#)或传代之间(4#→9#)均未检测出明显的遗传特征突变基因,其他表象正常受检者之间亦是如此,检测结果中唯一具有遗传致病性基因为8#的GJB2基因,经分析该基因与先天性耳聋疾病相关[5],而与本案PJS疾病相关的STK11基因突变在6位受检者中均为检出,究其原因发现用传统的突变检测方法难以有效检测出STK11基因突变情况,因此作者改用MLPA方法检测STK11基因的大片段缺失,该方法可在一次反应体系中快速、敏感、高效地同时检测出多达40个目的片段的相对拷贝数[6]。在本案例家族体系中,3例血液样本(6#,10#,11#)和3例唾液样本(4#、8#、9#)送至北京泛生子医学检验所有限公司进行MLPA技术检测,检测结果显示4#、6#、9#均检测出STK11基因Exon2-3缺失(见图J、L),而家系中表型正常的成员未检出该缺失突变(见图K、M),与遗传图谱相表象相对应,提示此家族性PJS与胚系来源的STK11基因Exon2-3缺失突变密切相关。
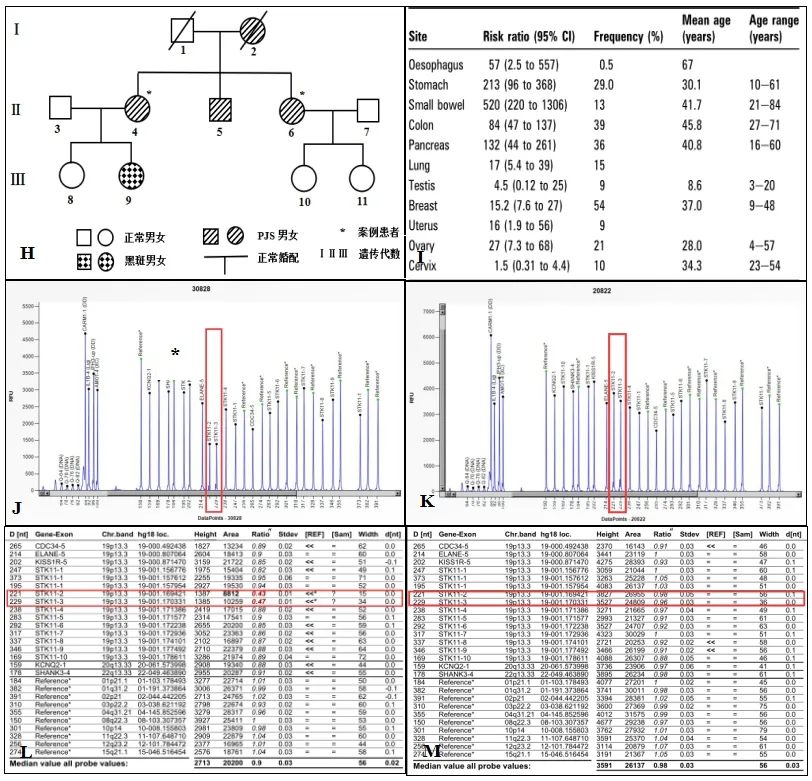
图H:患者家族遗传图谱(6#案例1,4#案例2);图I:PJS患者患癌风险;图J、L:STK11基因2号,3号外显子缺失(4#,6#,9#);图K、M:STK11基因正常(8#,10#,11#)
目前在我国人群中STK11缺失突变研究的报道较少,一方面受缺失突变分析方法和技术限制,另一方面存在有非STK11基因突变致病的PJS患者,Connolly等[7]在8例PJS患者的基因中却未发现STK11基因编码区的体细胞突变,而由STK11基因Exon2、3缺失突变导致的PJS也并非首次报道,Aretz等[8]采用MLPA方法从71例PJS患者中检测了17例STK11缺失,其中包括1例Exon2-3缺失;De Rosa M等[9]也报道了1例Exon2-3缺失的PJS患者;Nicoletta Resta等[10]在15例表现良好的PJS患者中检测到2例STK11 Exon2-3缺失,且其中1例尚未出现黑色素斑,进一步证实对本家族PJS诊断的正确性,也达到了基因检测的目的。
尽管PJS患者最常见的表现为胃肠道息肉,恶变率较低,但其发生肠癌的风险仍高于正常人群,且发生乳腺癌、子宫癌(宫颈癌)、胰腺癌等恶性肿瘤的风险也较正常人群高(如图I所示),且易伴发多器官、多系统的肿瘤是P-J综合征的重要特点。本家系合计4例为PJS患者,其中女性3例,2例发现宫颈腺癌,符合PJS较高肿瘤易感性的特点。此外,患者的母亲死因为子宫恶性肿瘤,并有皮肤黏膜黑斑表现,也进一步佐证了PJS易并发恶性肿瘤的遗传性特征。
PJS伴宫颈腺癌比较罕见,多见于个案报道,因随访时间较短,预后尚不明确。本案2例患者诊断时均为中晚期,例1患者术前相关检查均为阴性结果,可见PJS患者利用常规的宫颈筛查手段难以早期诊断。因此,对于PJS患者,能尽早严密随访至关重要。美国NCCN指南推荐,PJS患者每年行盆腔检查及子宫颈细胞学检查开始的年龄应为18~20岁,比普通人群开始筛查的年龄更早。对于医师而言,应了解PJS与妇科恶性肿瘤发生风险的关系,建议PJS患者在18岁后每年进行盆腔检查及子宫颈细胞学检查,对患者家属推荐在不同的年龄段进行多种肿瘤的筛查,以达到早诊断、早治疗的目的。
