长链非编码RNA在痛风性关节炎研究中的新进展
2020-05-14何欣黄玉琴陈炳利林福胜张全波
何欣,黄玉琴,陈炳利,林福胜,张全波
(1.川北医学院,四川 南充 637000;2.川北医学院附属医院老年医学科,四川 南充 637000)
痛风是嘌呤代谢紊乱和(或)尿酸排泄减少导致血尿酸升高,单钠尿酸盐(monosodiumurate,MSU)晶体沉积在关节、皮下、肾脏等引起的一组慢性疾病,主要表现为急性痛风性关节炎。痛风性关节炎是最常见的炎性关节炎,在过去几十年,痛风发病率呈显著升高趋势,痛风引起的急性疼痛、功能障碍和永久性关节损伤,给患者带来了巨大的身心伤害及沉重的经济负担,并且痛风还与许多慢性疾病(糖尿病、脑卒中、肾脏病、高血压、肥胖等)相关[1]。痛风的免疫学发病机制目前仍不明确,因此,探索痛风的发生、发展对指导临床诊断、缓解临床症状及用药、改善预后、减轻社会负担具有重大意义。长链非编码RNA(long non-coding RNAs,lncRNA)具有多种生物学功能。慢性炎症参与许多疾病的发生和发展,包括肥胖、动脉粥样硬化、骨关节炎、自身免疫性和退行性疾病。炎症过程由趋化因子、细胞因子和不同的炎症细胞介导,而非编码RNA在炎症反应的进展和管理中可能发挥的作用。目前,关于lncRNA参与炎症介导的过程以及与代谢紊乱相关的稳态失衡的研究结论较少,但研究发现,lncRNA可作为转录和转录后修饰及翻译的调节因子[2]。近年来,随着新的lncRNA及信号通路在痛风中被发现,可能为临床疾病诊断及治疗提供新思路。现就lncRNA在痛风性关节炎研究的新进展予以综述。
1 lncRNA来源、分类、与免疫学相关疾病
1.1lncRNA来源 lncRNA是长度超过200个核苷酸的非编码RNA分子序列,在进化中具有高度保守性。lncRNA与信使RNA结构相似,由RNA聚合酶Ⅱ转录而来,包括5′端帽子结构和3′端多聚腺苷酸尾,但是lncRNA缺乏开放的阅读框,不具有编码蛋白质的能力[2]。lncRNA主要分布于细胞核,少量分布在细胞质中。
1.2lncRNA分类 根据lncRNA的位置分为基因内lncRNA、基因间lncRNA(long intergenic non-coding RNA,lincRNA)、增强子RNA、双向lncRNA及反义lncRNA,见表1[3]。
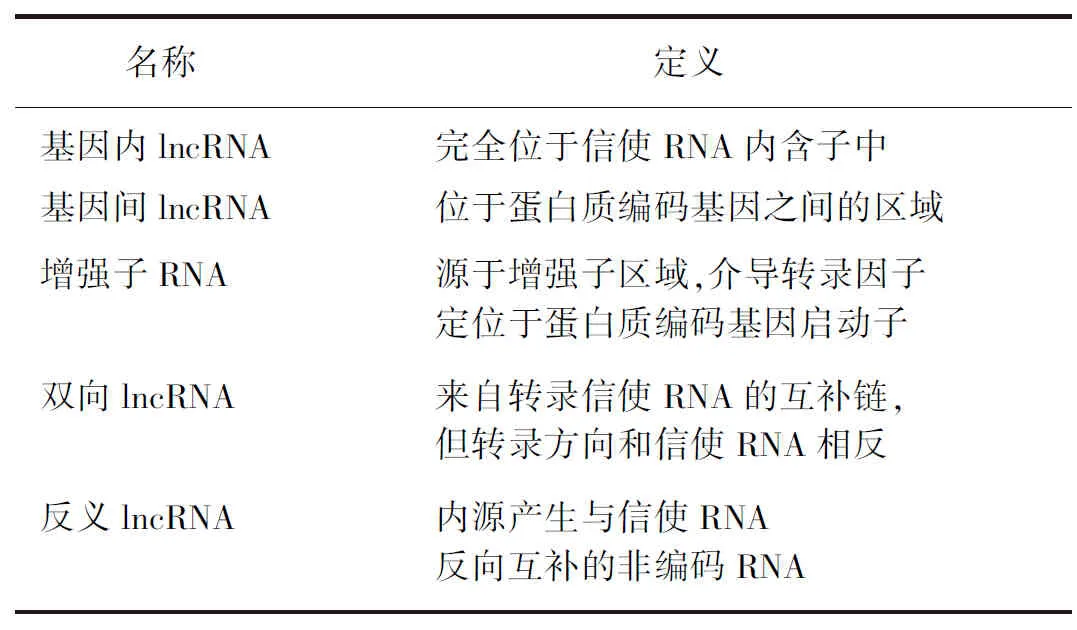
表1 lncRNA的分类
lncRNA:长链非编码RNA
1.3lncRNA参与的免疫学相关疾病 lncRNA因不编码蛋白质,在过去的基因-信使RNA-蛋白质-功能的研究模式中,lncRNA曾经被认为是基因转录过程的“垃圾”,但后续的研究发现lncRNA尽管没有被翻译成蛋白质,但是它确是功能分子[4]。lncRNA与其他分子结合参与了许多细胞过程,如染色体结构调节、转录、剪接、信使RNA的稳定性调节、信使RNA的可用性和翻译后修饰以及lncRNA根据其序列和二级结构为DNA、信使RNA、蛋白质提供相互作用域等[2]。非编码RNA在炎症反应的进展和管理中可能发挥作用,一些非编码RNA被证明是炎症相关介质的重要转录调控因子,如微RNA(microRNA,miRNA)-126、miR-132和miR-146,这些非编码RNA正作为具有诊断价值的生物标志物,在预后预测、个性化治疗中发挥指导作用[4]。lncRNA在各种生物学和免疫学过程中起着关键作用,参与了机体固有免疫和适应性免疫,有证据支持lncRNA参与自身免疫性疾病和炎症性疾病的发病机制[5]。如lincRNA-环氧合酶2(cyclooxygenase 2,COX2)已经被证明可以调节先天免疫细胞免疫基因的激活和抑制;树突状细胞(dendritic cells,DCs)lncRNA在DCs分化、抗原表达和免疫激活中起着重要作用;在免疫相关性疾病研究中发现,生长阻滞特异性转录因子5(growth arrest-specific transcript 5,GAS5)是系统性红斑狼疮的易感基因;类风湿关节炎与Hotair表达相关;多发性硬化与lnc-MAF4(MADS affecting flowering 4)相关[6]。然而,lncRNA在痛风的研究中目前较少,进一步研究lncRNA与痛风的关系可以帮助更好地了解痛风免疫学发病机制,并提供新的药物靶点和治疗方向。
2 痛风的免疫学发病机制
痛风是嘌呤代谢紊乱和(或)尿酸排泄减少导致血尿酸升高,MSU与局部微环境相互作用诱发的炎症反应。血中尿酸水平升高超过溶解度,析出并沉积关节软骨周围的MSU,诱导白细胞趋化聚集,活化固有免疫细胞,进而启动机体的固有免疫系统。参与的免疫细胞主要包括巨噬细胞、DC、中性粒细胞等。痛风与固有免疫相互作用机制尚不完全明确,可能涉及多条与炎症相关的信号通路。
2.1核因子κB(nuclear factor-kappa B,NF-κB)信号通路 Toll样受体(Toll like receptors,TLR)是位于细胞膜表面的模式识别受体,参与固有免疫,也是将固有免疫和适应性免疫连接起来的桥梁。危险信号MSU可识别TLR,活化的TLR通过髓样分化因子88(myeloid differentiation factor 88,MyD88)依赖通路转导信号,结合并活化白细胞介素(interleukin,IL)-1受体相关激酶,随后结合转化生长因子受体关联因子6,最终激活NF-κB[7]。活化的NF-κB能进入细胞核,完成相关炎性基因的转录,产生IL-1β前体(pro-IL-1β)。
2.2核苷酸结合寡聚化结构域样受体蛋白3(nuc-leotid binding oligomerization domain-like receptor protein 3,NLRP3)炎性小体信号通路 NLRP3为在巨噬细胞和DC胞质中存在的多蛋白复合物,属于NOD样受体(NOD-like receptors,NLRs)家族,是细胞质中的模式识别受体,在炎症性疾病中发挥重要作用。MSU可被NLRP3识别,NLRP3活化后结构发生改变,随后募集凋亡相关点样蛋白,并介导不活跃的胱天蛋白酶前体(pro-caspase-1)募集形成复合物,即NLRP3炎性小体;pro-caspase-1裂解为其活性形式,活化后的caspase-1能将无活性的IL-1β和IL-18前体剪切为成熟的IL-1β和IL-18,并分泌相应的活性成分至细胞外,引起炎症反应[8]。
2.3腺苷三磷酸(adenosine triphosphate,ATP)-嘌呤能离子通道型受体7(purinergic receptor P2X ligand-gated ion channel 7,P2X7R)信号通路 TLR及NLR信号通路在MSU诱发IL-1β分泌进而参与痛风的发病中起重要作用,但无法解释部分高尿酸血症终身不发展为痛风的现象,提示可能有MSU以外的致病途径参与了IL-1β的分泌。已有研究表明,ATP可作为刺激信号刺激P2X7R诱导IL-1β分泌[9]。痛风发作的主要诱发因素包括剧烈运动、感冒、酗酒和暴饮暴食,它们都有一个共同的特征,即体内ATP发生了剧烈变化,表明ATP的变化可能是急性痛风性关节炎的第二个致病信号。痛风患者外周血中CD14+P2X7R+细胞百分比显著高于高尿酸血症患者,可能解释了部分高尿酸血症不发展为痛风[10]。研究表明,与P2X7R功能相关的单核苷酸多态性可能与痛风性关节炎有关,说明ATP-P2X7R信号通路在痛风的发病中起着重要的调控作用[11]。
3 lncRNA对痛风发病通路的调节
目前lncRNA在痛风的发病中研究较少,具体机制并不清楚。但lncRNA可能参与了痛风的发病,姚承佼等[12]发现,多条lncRNA在痛风患者中异常表达,可能参与痛风的发生、发展,但具体机制仍不清楚;Lee等[13]发现,MSU诱导破骨细胞分化过程中lncRNA Janus激酶3差异表达,并揭示了lncRNA-Janus激酶3在通过Janus激酶3/活化T细胞核因子1(nuclear factor of activated T-cells 1,Nfatc1)/组织蛋白酶K轴分化在破骨细胞中起着关键作用,为治疗痛风性关节炎提供新思路。进一步研究lncRNA 与痛风发病的关系将为痛风的诊治提供新思路。
3.1lncRNA与NF-κB信号通路
3.1.1lncRNA与TLR 核富集转录本1(nuclear enriched abundant transcripts 1,NEAT1)是细胞核内的lncRNA,研究提示,NEAT1参与TLR2介导的先天免疫调节,并且选择性地调控一组TLR2信号活化缓慢持续诱导的晚期反应炎症因子的表达[14],参与TLR2信号通路的正负反馈调节;NEAT1与let-7a相互作用调节TLR4,上调炎症反应水平[15]。同时发现,lincRNA DYN-LRB2-2负调控TLR2表达[16]。乳头状甲状腺癌易感侯选物3(papillary thyroid carcinoma susceptibility candidate 3,PTCSC3)为lncRNA,正向调节TLR4的表达[17]。母系表达基因3(maternal expression gene 3,MEG3)为位于染色体14q32,由10个外显子组成的lncRNA,正向调控TLR4,促进炎症的发生[18]。lncRNA肺癌转移相关转录本1(metastasis-associated lung adenocarcinoma transcript 1,MALAT1)可以调节TLR4-NF-κB轴[19]。
3.1.2lncRNA与MyD88 MyD88是TLR4通路的关键适配器蛋白,可启动下游炎症因子的转导。miR-223在类风湿关节炎患者中被发现升高,同时被证实与破骨细胞形成和骨侵蚀有关[20]。研究发现,miR-223通过直接靶向信号转导及转录激活因子3,增强小鼠巨噬细胞中IL-6和IL-1β的产生[21]。研究表明,被转化生长因子-β激活的lncRNA(lncRNA activated by transforming growth factor β,lncRNA-ATB)可通过抑制miR-223,阻断MyD88/NF-κB通路,减轻炎症反应,但miR-223与lncRNA-ATB机制目前仍不明确,需要进一步研究[22]。研究显示,lncRNA MALAT1可提高MyD88启动子H3乙酰化水平,而正向调控MyD88的表达[23]。
3.1.3lncRNA与NF-κB NF-κB是一种转录因子,在调控炎症反应、免疫功能、恶性转化中发挥重要作用。NF-κB信号通路的异常活动可能导致炎症、自身免疫性疾病和肿瘤发生。已鉴定出NF-κB家族中的6个转录因子,即RelA(p65)、RelB、c-Rel、p50(p105前体)、p52(p100前体)和Relish,痛风患者在静息状态时,NF-κB以无活性形式存在,当刺激信号刺激时,活化的IκB激酶磷酸化IκB蛋白导致其泛素化而降解,NF-κB二聚体释放入细胞核,激活下游靶基因转录,刺激生成pro-IL-1β[24]。lncRNA Lethe可以作为负反馈分子,通过与RelA结合,抑制NF-κB驱动的基因转录,Lethe的下调可增强NF-κB的表达;NF-κB相互作用的lncRNA(NF-kappa B interacting lncRNA,NKILA)通过能修饰 IκB的磷酸化序列,阻止IκB的降解,从而阻止NF-κB向细胞核易位,抑制NF-κB活化,抑制了NF-κB驱动的炎症;lncRNA PACER(p50-associated COX-2 extragenic RNA)可与NF-κB的抑制性亚基p50相互作用,螯合p50,可促进活化p65/p50二聚体的形成,促进炎症和免疫的重要介质环加氧酶-2的基因表达[25]。促进红细胞存活的lncRNA(lncRNA-erythroid-pro-survival,lincRNA-EPS)为红系细胞分化调节子,在TLR配体激活后EPS显著抑制,下调NF-κB,研究表明,在lncRNA-EPS缺陷的小鼠中容易产生炎症因子,也更容易发生致死性炎症[26]。以上lncRNA可通过调节NF-κB影响炎症的发生,但在痛风患者中研究甚少,需要后期进一步的研究。
3.2lncRNA与NALP3炎性小体 NLRP3炎性小体由多种异常多样的病原相关分子模式激活,包括一些病毒、细菌、真菌病原体、尿酸结晶、ATP等。在痛风患者中,MSU诱导NLRP3炎性小体异常高表达,诱发炎症反应,为痛风发病关键步骤,证据表明,NLRP3炎性小体的激活可以通过去泛素化机制调控[27]。研究已证实,去泛素化酶含BRCC3(BRCA1-BRCA2-containing complex subunit 3)能够降低NLRP3蛋白泛素化水平,促进NLRP3炎性小体的活化[28]。进一步研究这种泛素化修饰的调控机制,可能会发现调控NLRP3炎性小体活化的“分子开关”。INK4基因座中反义非编码RNA(antisense non coding RNA in the INK4 lucos,ANRIL)通过miR-122-5p上调BRCC3的表达,促进NLRP3炎性小体活化,促进IL-1β、IL-18、肿瘤坏死因子-α、IL-6等炎症细胞因子的分泌[28]。研究发现,lncRNA MALAT1可以正向调控NLRP3炎性小体的表达[29]。值得注意的是,Han等[30]描述了lncRNA MALATI包含一个miR-133功能靶点,并作为竞争性的内源性RNA抑制miR-133的作用。在之前的研究中,miR-133已被证明通过结合体内NLRP3的3′非翻译区而抑制NLRP3的表达[31],但仍需要对lncRNA、miRNA和NLRP3炎性小体之间的潜在联系进行系统研究。lncRNA NEAT1可促进NLRP3炎性小体的活化,并增强caspase-1活性,NEAT1与pro-caspase-1结合,促进NLRP3炎性小体的组装,并形成稳定成熟的caspase-1四聚体,增加caspase-1的蛋白酶活性,因此,NEAT1可能是NLRP3炎性小体相关疾病(如痛风和自身炎性综合征)的治疗靶点[32]。上述发现的一些lncRNA可能影响痛风发生、发展的过程,成为炎症控制及药物治疗的靶点,但仍需要更多进一步的研究。
3.3lncRNA与IL-1β IL-1β在急性痛风性关节炎发生过程中逐级放大炎症。lncRNA MEG3有10个转录亚型,其中的MEG3-4在动物实验中显示可以调节IL-1β的表达,MEG3-4过量可诱导IL-1β的表达[33]。MEG3-4有可能成为控制炎症因子释放的有效作用靶点。有研究发现,小核糖体管家基因RNA 1(small uncleolar RNA host gene 1,SNHG1)可以减轻IL-1β引起的关节炎,并且揭示SNHG1可能是骨关节炎诊断和治疗的潜在靶点[34]。研究发现,lncRNA ZBED3-AS1(zinc-finger BED domain containing 3-antisense 1)对IL-1β的表达有负性调控作用[35]。另外,有研究提示,氨甲酰磷酸合成酶1-内含子转录本1(carbamoyl-phosphate synthase 1-intronic transcript 1,CPS1-IT1)可减少IL-1β的表达,从而抑制NF-κB信号通路;脂多糖诱导的MALAT1激活IκB激酶β/NF-κB信号通路,通过下调miR-199b促进促炎症因子、肿瘤坏死因子-α和IL-1β的产生;更重要的是,MALAT1的下调抑制了肿瘤坏死因子-α、IL-1β,有望通过剔除MALAT1基因抑制炎症反应[36],但具体机制目前仍不明确,需要进一步的研究证实[37]。
3.4lncRNA与ATP-P2X7R信号通路 嘌呤能受体可分为两大类,即P1(腺苷)受体和P2(ATP)受体,P2受体可分为ATP门控通道和G蛋白偶联受体。P2X7R是离子门控P2X家族中重要的成员,该家族由595个氨基酸组成,其N端序列结构高度保守,与P2X受体家族其他成员具有高度同源性。P2X7R是ATP门控阳离子通道,广泛分布于人体的各种组织和器官中,主要在巨噬细胞、DC、淋巴细胞和肥大细胞中表达。P2X7R具有多种生理功能,尤其在介导炎症反应方面。与其他P2X受体相比,P2X7R与ATP的亲和力较弱,在组织损伤和感染引发炎症的情况下,ATP可被激活。P2X7R是P2XRs家族中与炎症反应最相关的嘌呤受体,P2X7R在炎症状态下表达显著上调,激活多种细胞内信号转导通路,调节炎症介质的释放,参与免疫反应和炎症反应,诱导细胞损伤甚至凋亡,最终导致免疫性疾病[38]。UC.48+是来自人、小鼠、大鼠基因组的lncRNA,研究提示,UC.48+是P2X7R的靶基因,沉默的UC.48+降低P2X7R表达,阻止NF-κB的表达,UC.48+表达增强,P2X7R的表达也显著增加,并激活了NF-κB,影响炎症的发生[39]。但目前关于UC.48+研究报道较少,可能需要更多的研究了解其作用机制。P2X7R广泛表达于人体肠道和免疫细胞中,是机体抵抗病原体侵袭的重要组成部分,但目前研究较少,进一步的深入研究,可能为炎症性疾病的治疗提供新的靶点。
4 小 结
痛风是最常见的炎性关节炎,与糖尿病、心血管疾病、肾脏病等慢性病相关,给患者带来了严重的经济和心理负担。随着基因组研究的深入,越来越多研究表明lncRNA 参与人类免疫、炎症、肿瘤性疾病的发生、发展。例如,尿液中的前列腺癌抗原3已作为前列腺癌辅助诊断指标[40];在类风湿研究中发现,甲氨蝶呤通过DNA活化蛋白激酶催化亚基的依赖机制诱导lincRNA-p21产生,继而抑制NF-κB活性,达到抗炎的目的[12]。目前,痛风发病的机制尚不完全明确,lncRNA在痛风中的研究报道也较少。因此,进一步研究痛风发病机制及可能的参与调节痛风性关节炎的lncRNA,有望在将来成为疾病诊断的标志物及治疗靶点。
