Cutaneous nodules and a novel GNAS mutation in a Chinese boy with pseudohypoparathyroidism type Ia: A case report and review of literature
2020-05-14
Yun-Ling Li, Department of Dermatology, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou 310052, Zhejiang Province, China
Ting Han, Department of Children’s Health Care, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou 310052,Zhejiang Province, China
Fang Hong, Department of Genetics and Metabolism, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou 310052, Zhejiang Province, China
Abstract
BACKGROUND
Pseudohypoparathyroidism type Ia (PHP Ia) is a rare hereditary syndrome, and patients with early PHP Ia are generally not diagnosed based on the presentation of cutaneous nodules as the main clinical feature. Here, we describe a Chinese boy with PHP Ia in whom the main clinical feature was cutaneous nodules, and the patient exhibited a novel GNAS mutation.
CASE SUMMARY
A 5-year-old boy presented with a 5-year history of cutaneous nodules scattered over his entire body. The patient had a short stature, round face, short neck, and slightly flattened nose; he also had multiple hard papules and cutaneous nodules scattered over his entire body. The patient had a significantly elevated parathyroid hormone level. His serum calcium level was reduced, while his serum phosphorus level was increased and his serum thyroid-stimulating hormone level was elevated. Skin biopsy showed osteoma cutis in subcutaneous tissue. Sanger sequencing revealed a frameshift mutation, c.399delT(p.Ser133Argfs*2) in exon 5 of the GNAS gene. The patient was diagnosed with PHP Ia and subclinical hypothyroidism. He was given 1,25-dihydroxyvitamin D,calcium carbonate, and synthetic L-thyroxine. After 3 months of treatment, the patient’s parathyroid hormone level decreased, and his serum calcium and serum phosphorus levels were normal. Moreover, his thyroid-stimulating hormone level decreased.
CONCLUSION
These findings can help dermatologists to diagnose PHP Ia in patients with cutaneous nodules as the main early clinical feature.
Key words: Pseudohypoparathyroidism type Ia; Cutaneous nodules; GNAS;Pseudohypoparathyroidism; Case report
INTRODUCTION
Pseudohypoparathyroidism (PHP) is a rare, sporadic or familial disorder characterized by an elevated plasma level of parathyroid hormone (PTH) caused by resistance to PTH action in target tissues[1,2]. PHP was first reported in 1942 by Albrightet al[3]and classified into PHP type I (PHP I) and PHP type II (PHP II)according to the renal response to exogenous PTH administration. PHP I is divided into subtypes Ia, Ib, and Ic. Patients with PHP Ia display hypocalcemia, hyperphosphatemia, PTH resistance, and variable developmental abnormalities [e.g.,Albright’s hereditary osteodystrophy (AHO)]. PHP Ia is caused by inactivating mutations of the guanine nucleotide-binding protein, alpha-stimulating activity polypeptide (GNAS) gene, which encodes the alpha subunit of the stimulatory heterotrimeric G protein (Gsα)[4].
Patients with PHP Ia generally seek medical attention due to epileptic seizures, and both the misdiagnosis and missed diagnosis rates are extremely high. Here, we describe a patient with PHP Ia who presented with multiple cutaneous nodules as the main clinical feature; this patient did not experience epileptic seizures and exhibited a novelGNASmutation.
CASE PRESENTATION
Chief complaints
A 5-year-old boy with a 5-year history of cutaneous nodules was referred to the Children’s Hospital at Zhejiang University School of Medicine, Hangzhou, China.
History of present illness
Cutaneous nodules had been observed since the patient was 2 mo old. A hard papule and nodules were first found at the waist. Numerous similar lesions subsequently appeared on the head, trunk, and limbs; these gradually became progressively larger.
History of past illness
The patient had no significant medical history, psychiatric history, or history of substance misuse.
Personal and family history
The patient was born at a gestational age of 34 wk with a birth weight of 2.6 kg and height of 46 cm. The patient’s weight was between the 6thand 10thpercentiles from birth to the age of 3 mo. His weight then began to increase rapidly such that obesity was evident at the age of 4 mo (Figure 1A) and continued until the age of 9 mo (> 97thpercentile). The patient’s height was between the 6thand 10thpercentiles from birth to the time of this report. The patient could raise his head at the age of 6 mo, sit up at the age of 1 year, and walk at the age of 2 years. The patient’s mother had exhibited similar cutaneous nodules in the calf and trunk regions at the age of 26 years; she also had a short stature, round face, and was obese (Figure 1D). The patient’s older brother, father, aunt, and uncle did not display similar manifestations of the disease.
Physical examination
On physical examination, the child had a short stature, round face, short neck, and slightly flattened nose (Figure 1B and C). His height was 105 cm and his weight was 19.5 kg (10thand 75thpercentiles, respectively). The patient was only able to express simple sentences; his score on the Wechsler Intelligence Scale for Children was 51,which suggested mental retardation. The patient had multiple hard papules and cutaneous nodules scattered over his entire body; these cutaneous nodules measured 1 mm to 2 cm in size (Figure 2A). He exhibited no erythema, blisters, tenderness, or swelling on the surfaces of cutaneous nodules.
Laboratory examinations
Laboratory investigations showed normal findings in routine blood, urine, and stool tests, along with normal liver and renal function. The patient also had normal cortisol,adrenocorticotropic hormone, bone type alkaline phosphatase, and 25-hydroxyvitamin D levels. His serum calcium level was 2.03 mmol/L (normal range,2.20-2.65 mmol/L) and his serum phosphorus level was 2.91 mmol/L (normal range,1.29-2.26 mmol/L); his serum PTH level was significantly elevated (466.2 ng/L;normal range, 15-65 ng/L). The patient had an elevated thyroid-stimulating hormone(TSH) level of 12.34 mIU/L (normal range, 0.35-4.94 mIU/L) with a normal thyroxine level of 71.87 nmol/L (normal range, 62.68-150.80 nmol/L) and a normal triiodothyronine level of 2.01 nmol/L (normal range, 0.88-2.44 nmol/L). A skin biopsy from his right thigh showed heterotopic ossification within the subcutaneous tissue,indicative of osteoma cutis (Figure 2B). Sanger sequencing showed that a c.399delT heterozygous gene mutation (NM_000516.4) was present in exon 5 ofGNASin both the patient and his mother.
Imaging examinations
Brain computed tomography examination showed multiple calcium-like high-density calcifications in the patient’s scalp, but not within the brain. Electroencephalogram showed no seizure-like waves.
FINAL DIAGNOSIS
The proband was diagnosed with PHP Ia by the presence of hormone resistance,cutaneous ossification, obesity, and AHO phenotype, and maternal inheritance of theGNASmutation. This proband had elevated TSH levels and normal T3 and T4 levels,so a diagnosis of subclinical hypothyroidism was made. Therefore, the final diagnosis of the patient was PHP Ia combined with subclinical hypothyroidism.
TREATMENT
The patient was given 1,25-dihydroxyvitamin D at a dose of 0.25 μg once daily and calcium carbonate at a dose of 300 mg once daily; he was instructed to avoid dairy,soy, and other high-phosphorus food products. Because the patient’s subclinical hypothyroidism led to concerns regarding his neurologic development, he was also administered synthetic L-thyroxine at a dose of 12.5 μg once daily.
OUTCOME AND FOLLOW-UP
After 3 mo of treatment, the patient’s PTH level decreased to 184.10 ng/L (normal range, 15-65 ng/L) with serum calcium and phosphorus levels of 2.24 (normal range,2.20-2.65 mmol/L) and 2.05 mmol/L (normal range, 1.29-2.26 mmol/L), respectively;his TSH level decreased to 8.12 mIU/L (normal range, 0.35-4.94 mIU/L).
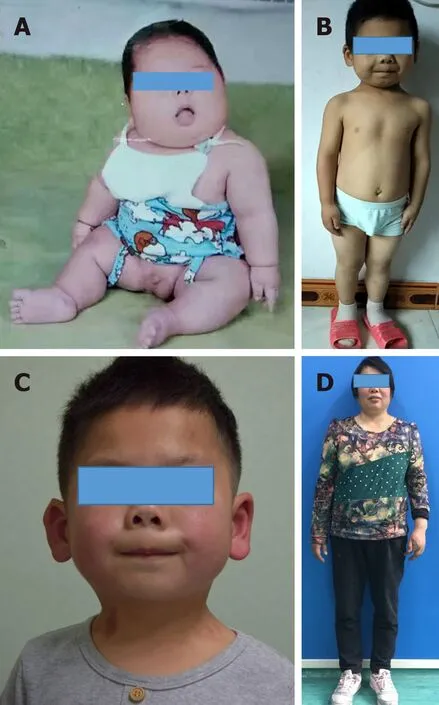
Figure 1 Photographs of the patient and his mother. A-C: The patient had severe obesity at 4 mo old (A) and a round face, short neck, flat nose at 5 years and 1 mo old (B and C); D: His mother is short and obese with a round face.
DISCUSSION
PHP is a rare autosomal dominant inherited disorder that is associated with an exon mutation in the maternal allele of theGNASgene. TheGNASgene is located on chromosome 20q13, which consists of 13 exons and 12 introns. The product of theGNASgene is Gsα, which is an important component of the cAMP/protein kinase.PTH mainly couples with Gsα through the PTH receptor to form a complex that activates adenylate cyclase and promotes the generation of cAMP, thereby regulating the cell response.GNASis a complex imprinted gene encoding Gsα that exhibit exclusively maternal or paternal expression[5]. Maternal allele of theGNASgene is the only source of Gsα in the kidney (the paternal allele is normally silenced in this tissue). In PHP Ia, mutations in the maternalGNASallele result in a marked reduction of Gsα levels, leading to failure to elicit an appropriate increment in urinary cAMP and phosphate excretion following exogenous PTH infusion and PTH resistance[6].Because the target organs (i.e., the kidney and bone) do not respond to PTH,hypocalcemia feedback stimulates excessive parathyroid secretion of PTH.
The clinical manifestations of PHP Ia include signs of hypoparathyroidism,characterized by calcium and phosphorus metabolism disorders[7-9]. Some patients also demonstrate a typical AHO phenotype, including short stature, obesity, round face, short neck, short and coarse fingers/toes, and ectopic ossification. The molecular mechanisms ofGNAS-related heterotopic ossification have not been fully elucidated.It might involve aberrant differentiation of mesenchymal progenitor cells in the dermis or subcutaneous fat. Researchers have reported that activation of Hedgehog signaling causesGNAS-related heterotopic ossification. In PHP Ia with mutations inGNAS, Hedgehog signaling is activated in progenitor cells, which leads to heterotopic ossification and inhibition of formation of adipocytes[10,11]. Heterotopic calcification or ossification is extremely common in patients with AHO, which mainly occurs in the subcutaneous tissue and brain parenchyma; the most common manifestation is calcification of the brain basal ganglia. Seizures can also be caused by brain parenchymal calcification and hypocalcemia. Thyroid hormone, growth hormone,and gonadotropin are mediated by Gsα; therefore, patients with aGNASmutation may present with resistance to other hormones, including TSH, growth hormone, and gonadotropin-releasing hormone. Therefore, patients with PHP Ia may exhibit hypothyroidism and hypogonadism. The patient in this case had an elevated TSH level and normal triiodothyronine and thyroxine levels; thus, a diagnosis of subclinical hypothyroidism was made.
PHP Ia needs to be distinguished from PHP Ib, PTH Ic, and pseudo-PHP. PHP type Ib is a rare genomic disease that is typically sporadic; a minority of patients demonstrate autosomal dominant inheritance. PHP Ib is associated with abnormal epigenetic regulation of theGNASgene due to the loss ofGNASpromoter methylation; theGNASgene coding sequence is unaltered. In addition to PTH resistance, the clinical manifestations of PHP Ib typically include resistance to many other hormones. PHP Ib is generally not accompanied by AHO characteristics, but some patients may exhibit short fingers and toes. Patients with PTH Ic exhibit AHO characteristics and resistance to multiple hormones, but have normal Gsα activity;thus far, the molecular and genetic mechanisms of PTH Ic remain unknown. Pseudo-PHP is a syndrome caused by a mutation of the paternal allele of theGNASgene;affected patients exhibit AHO characteristics, as well as normal serum calcium and normal serum phosphorus, but lack PTH resistance.
There have been previous reports of patients with PHP Ia in whom the diagnosis was confirmed by genetic analysis. PHP Ia cases withGNASmutations previously described are reviewed[12-33]and summarized in Table 1. For example, two Korean patients with PHP Ia were confirmed to have a nonsense mutation of c.94A>T(p.Lys32X) and a known frameshift mutation of c.344_345insT (p.V117RfsX23) in theGNASgene[24]. Another study of patients with PHP Ia revealed two novel mutations:A c.569_570del mutation (p.Tyr190CysfsX19) and a splicing mutation (c.659+1G>A) in theGNASgene[25]. A Turkish boy was reported to have PHP type Ia (GNASgene mutation, IVS4+5G>C); his mother had pseudo-PHP[33]. Here, we describe a 5-year-old boy with PHP Ia who had the c.399delT (p.Ser133Argfs*2) mutation in theGNASgene; his mother carried the same mutation. Notably, the C.399delT mutation of exon 5 in theGNASgene is a frameshift mutation; this frameshift mutation causes AGT→AGG, resulting in an Arg to Ser amino acid variant. The second codon after this mutation site (c.399delT) becomes the stop codon (TGA), which induces early termination of translation and leads to a truncated protein associated with partial PTH resistance. We searched for this mutation in published online databases including Exome Aggregation Consortium, genome Aggregation Database, ClinVar database, and Human Gene Mutation Database; there was no information about the site in these databases. Therefore, we concluded that the c.399delT mutation in theGNASgene is novel.
CONCLUSION
PHP Ia is a rare genetic disease with an extremely high rate of missed diagnosis. The findings in this report can help dermatologists to recognize and accurately diagnose PHP Ia in patients with cutaneous nodules as the main early clinical feature. If necessary, genetic testing should be conducted to confirm the diagnosis.
杂志排行

World Journal of Clinical Cases的其它文章
- Comprehensive review into the challenges of gastrointestinal tumors in the Gulf and Levant countries
- Can cyclin-dependent kinase 4/6 inhibitors convert inoperable breast cancer relapse to operability? A case report
- Ruptured splenic peliosis in a patient with no comorbidity: A case report
- Successful kidney transplantation from an expanded criteria donor with long-term extracorporeal membrane oxygenation treatment: A case report
- Boarding issue in a commercial flight for patients with cavitary pulmonary tuberculosis: A case report
- Cytomegalovirus ileo-pancolitis presenting as toxic megacolon in an immunocompetent patient: A case report