磁性表面分子印迹聚合物(Fe3O4@mSiO2@MIP)的制备及四溴双酚A检测研究
2020-05-13费贵强邵彦明王海花李璐璐
费贵强, 白 浩, 邵彦明, 王海花, 李璐璐
(陕西科技大学 化学与化工学院 陕西省轻化工助剂重点实验室, 陕西 西安 710021)
0 引言
四溴双酚A(TBBPA),作为最常用的溴代阻燃剂之一,已经在各种塑料和电子产品中被使用[1].但是它也被视为一种持久性有机污染物,已经存在于污水、野生动植物和人类血清中[2,3].所以有效地检测环境中微量TBBPA具有非常重要的意义.
传统的检测方法例如气相色谱串联质谱(GC-MS),高效液相色谱串联质谱(HPLC-MS),固相萃取和其他复杂的技术通常用于TBBPA的检测和分析[4-8].但是由于环境中的TBBPA浓度相对较低以及所处环境的复杂性,基于色谱检测的传统检测技术通常存在仪器昂贵、成本高、样品处理复杂等弊端,从而限制了其广泛应用[9].因此需要一种简便且快速的检测技术用于TBBPA的检测和分离.
表面分子印迹技术(SMIP)是一种将分子印迹聚合物的识别位点固定在载体表面的印迹技术.由此制得的印迹聚合物具有更容易获得的结合位点,更快的传质速率和结合动力学[10,11].有效地避免了传统方法制备的印迹聚合物识别位点不易接近,模板分子难以洗脱等缺点.
活性可控自由基聚合综合了自由基聚合和离子聚合的优点,避免了传统自由基聚合在制备分子印迹聚合物时链增长速度不稳定的缺点,大大提高了聚合反应和聚合产物的结构可控性[12].作为其中之一的RAFT聚合不仅有着反应条件温和的优点,而且具有较宽的单体选择范围[13].为制备具有特定结构、超薄印迹层的分子印迹聚合物提供了可靠的技术.
本文结合了表面分子印迹技术和RAFT聚合的优势,使用Fe3O4纳米粒子作为内核,通过溶胶凝胶法包覆SiO2得到Fe3O4@SiO2.然后在十六烷基三甲基溴化铵的作用下,制备介孔Fe3O4@mSiO2并将其作为载体,通过4-氯甲基苯基三氯硅烷修饰并引入RAFT试剂,最后通过引发RAFT聚合制备了Fe3O4@mSiO2@MIP微球.所制备的印迹聚合物对TBBPA表现出良好的吸附能力,并且还能够在复杂的环境中抵抗其结构类似物的影响特异性识别TBBPA.与此同时,由于磁核的作用,印迹聚合物可以通过外加磁场被迅速地分离,表现出良好的重复使用性.
1 实验部分
1.1 实验原料
无水三氯化铁(FeCl3),双酚A(BPA),4-乙烯基吡啶(4-VP),4,4-联苯二酚(BIP),正硅酸乙酯(TEOS),对叔丁基苯酚(BP),三乙醇胺(TEA),四溴双酚A(TBBPA),AR,萨恩化学技术有限公司;4-氯甲基苯基三氯硅烷,十六烷基三甲基溴化铵(CTAB),三乙胺,偶氮二异丁腈(AIBN),二硫化碳(CS2),乙二醇二甲基丙烯酸酯(EGDMA),AR,天津市天力化学试剂有限公司.
1.2 Fe3O4@mSiO2纳米粒子的制备
参考文献[14]报道的方法制备单分散性Fe3O4纳米粒子.然后将0.2 g纳米Fe3O4在超声作用下均匀分散于80 mL无水乙醇和20 mL去离子水的混合溶液中,加入2.4 mL NH3·H2O,继续超声1 h,然后滴加0.2 mL TEOS于上述反应液中,并在室温下剧烈搅拌4 h.所得产物用磁铁分离,经过水和乙醇洗涤数次,并在真空下干燥,即可得到Fe3O4@SiO2纳米颗粒.
在超声作用下将0.15 g Fe3O4@SiO2纳米粒子分散于125 mL去离子水中.然后,向其中加入0.15 g CTAB和1.25 mL NaOH水溶液(0.1 mol/L),搅拌均匀后,滴加1 mL TEOS于上述分散液中.然后,将反应物在N2保护下在60 ℃的油浴中反应12 h.所得产物用磁铁分离,经过水和乙醇分别洗涤数次后,在真空条件下干燥.
将上述所得样品分散在10 mg/mL NH4NO3的乙醇溶液中,在80 ℃下每隔6 h更换溶剂,重复两次以完全地去除CTAB得到Fe3O4@mSiO2纳米粒子.
1.3 Fe3O4@mSiO2-RAFT纳米粒子的制备
首先在超声作用下将0.2 g Fe3O4@mSiO2分散于100 mL无水甲苯中,随后,向其中加入0.4 mL 4-氯甲基苯基三氯硅烷并搅拌均匀,缓慢滴加5 mL含有0.2 mL TEA的无水甲苯溶液到上述反应液中,并使体系保持在120 ℃下回流24 h.所得产物被磁铁分离后,经过水和乙醇分别洗涤数次后,在真空条件下干燥即得到Fe3O4@mSiO2-Cl.
将16 mL溴苯分散在120 mL无水的THF中,N2保护下,逐滴加入到装有3.4 g(0.14 mol)镁的250 mL三口烧瓶中,待反应发生后,加入剩下的混合液并开始搅拌,直至镁条消失.待反应结束后,向上述反应体系中加入9 mL(0.15 mol)CS2溶液,加热至40 ℃反应4 h.再向反应液中加入0.26 g Fe3O4@mSiO2-Cl纳米粒子,超声搅拌并分散均匀后升温至70 ℃反应72 h.产物被磁铁分离后,并用乙醇洗涤多次,真空环境下干燥,得到Fe3O4@mSiO2-RAFT纳米粒子.
1.4 Fe3O4@mSiO2@MIP纳米粒子的制备
将0.136 g TBBPA和0.21 mL的4-VP溶解于40 mL无水甲苯中,并在无氧环境下机械搅拌12 h后,超声分散40 mg Fe3O4@mSiO2-RAFT纳米粒子于上述溶液中.随后依次加入1.189 mL EGDMA和13 mg AIBN以引发聚合,使反应体系在N2保护下在60 ℃的油浴中反应24 h,之后磁分离产物,使用乙醇洗涤3次,并在真空下干燥.
用甲醇-乙酸混合液(9∶1,V∶V)作为洗脱剂,去除模板分子,直至洗脱剂中检测不到TBBPA.所得样品用乙醇洗至中性,磁分离后,真空条件下干燥即可得到含有印迹位点的Fe3O4@mSiO2@MIP纳米粒子.
采用与上述相同的方法制备了非印迹聚合物,即为Fe3O4@mSiO2@NIP纳米粒子,只是制备过程没有加模板分子TBBPA.
1.5 吸附性能
1.5.1 等温吸附实验
选用甲醇-水(1∶1,V∶V)作为溶剂,配置浓度为10 mg/L、20 mg/L、30 mg/L、40 mg/L、60 mg/L、80 mg/L、100 mg/L的TBBPA溶液.将5 mg Fe3O4@mSiO2@MIP或Fe3O4@mSiO2@NIP通过超声分散加入到含有5 mL不同浓度的TBBPA溶液中,并将其置于303 K、300 rpm的恒温摇床中振荡12 h.磁铁分离并通过HPLC确定溶液中TBBPA的浓度.
1.5.2 吸附动力学实验
将5 mg Fe3O4@mSiO2@MIP或Fe3O4@mSiO2@NIP超声分散于装有5 mL 40 mg/L的TBBPA水-甲醇溶液中,并在303 K、300 rpm的恒温摇床中分别振荡5 min、10 min、15 min、20 min、30 min、60 min、100 min,经磁铁分离吸附剂后,通过HPLC检测上清液中TBBPA的浓度.吸附剂对于TBBPA的吸附量(Qt),可通过公式(1)进行计算:
(1)
式(1)中:Ct为tmin后TBBPA的浓度(mg/L);C0为TBBPA的初始浓度(mg/L);m为加入Fe3O4@mSiO2@MIP或者Fe3O4@mSiO2@NIP的质量(g);V为TBBPA甲醇-水溶液的体积(mL).
1.5.3 选择性实验
BIP、BP和BPA被选择作为TBBPA的结构类似物来评价Fe3O4@mSiO2@MIP的选择性能.将5 mg Fe3O4@mSiO2@MIP、Fe3O4@mSiO2@NIP分别加入到50μmol/L的TBBPA、BIP、BP及BPA的甲醇-水溶液中,在相同于上述的条件下,将混合物在恒温摇床中振荡12 h,磁铁分离后,通过HPLC测定上清液中TBBPA及其结构类似物的浓度.
1.5.4 竞争性实验
将5 mg Fe3O4@mSiO2@MIPs加入到50μmol/L的TBBPA及其结构类似物(BPA、BIP、BP)的混合液中,相同条件下进行振荡.振荡结束后,使用HPLC测试各组分浓度.
1.5.5 色谱条件
采用反相C18色谱柱(150×4.6 mm,5μm),甲醇/水(V∶V=75∶25)被作为流动相,并且以1 mL/min的流速作为前提条件的情况下对各物质的含量进行测试.在波长为292 nm下对TBBPA进行检测,278 nm下检测BPA、BP和BIP.
2 结果与讨论
2.1 TEM表征
纳米材料的形貌通过TEM进行表征,其结果显示如图1所示.图1(a)为Fe3O4的TEM照片,可以看出,由水热法制备出的Fe3O4纳米颗粒显示出良好的单分散性,其粒径约为300 nm;从图1(b)可以看出,经过SiO2包覆之后,形成清晰的壳层结构,且Fe3O4纳米颗粒表面变得光滑,表明了Fe3O4@SiO2的成功制备;图1(c)为在CTAB作为致孔剂的条件下,包覆介孔SiO2之后形成的磁性介孔载体Fe3O4@mSiO2的TEM照片,与Fe3O4@SiO2纳米粒子相比,其壳层厚度进一步增加,并且从内插图中可以发现,壳层呈多孔状,说明具有丰富孔道结构的磁性介孔载体Fe3O4@mSiO2已经被成功制备;图1(d)为Fe3O4@mSiO2@MIP的TEM图,从图1中可以发现,通过表面引发的RAFT聚合在介孔载体上形成一层薄的聚合物层.

(a)Fe3O4

(b)Fe3O4@SiO2

(c)Fe3O4@SiO2@mSiO2

(d)Fe3O4@mSiO2@MIP图1 各阶段产物的TEM图
2.2 FTIR分析
每步反应阶段产物的结构均通过FTIR进行了表征,结果如图2所示.曲线a中450 cm-1、562 cm-1处的吸收峰归属于Fe3O4中Fe-O的振动吸收峰,1 400 cm-1、1 600 cm-1归属于-COOH的特征吸收峰,这可能来源于制备过程中加入的柠檬酸三钠[15];在曲线b中,出现了位于1 080 cm-1和785 cm-1处的特征峰,对应于Si-O-Si的伸缩振动和弯曲振动,同时Si-OH的伸缩振动吸收峰出现在944 cm-1处,说明在Fe3O4纳米粒子表面上形成了SiO2壳层[16];在曲线c中,670 cm-1处的微弱的吸收峰对应于C-Cl的特征吸收[17];并且在2 980 cm-1、2 867 cm-1出现了-CH3,CH2的吸收峰,说明卞氯结构被成功地引入到介孔载体表面;在曲线d中,1 122 cm-1处对应的C=S的振动吸收峰表明RAFT链转移剂被成功引入到磁性载体上;在曲线e中,1 610 cm-1和1 410 cm-1处的伸缩振动吸收峰匹配于吡啶环上的官能团,证明了Fe3O4@mSiO2@MIP的成功制备[18].
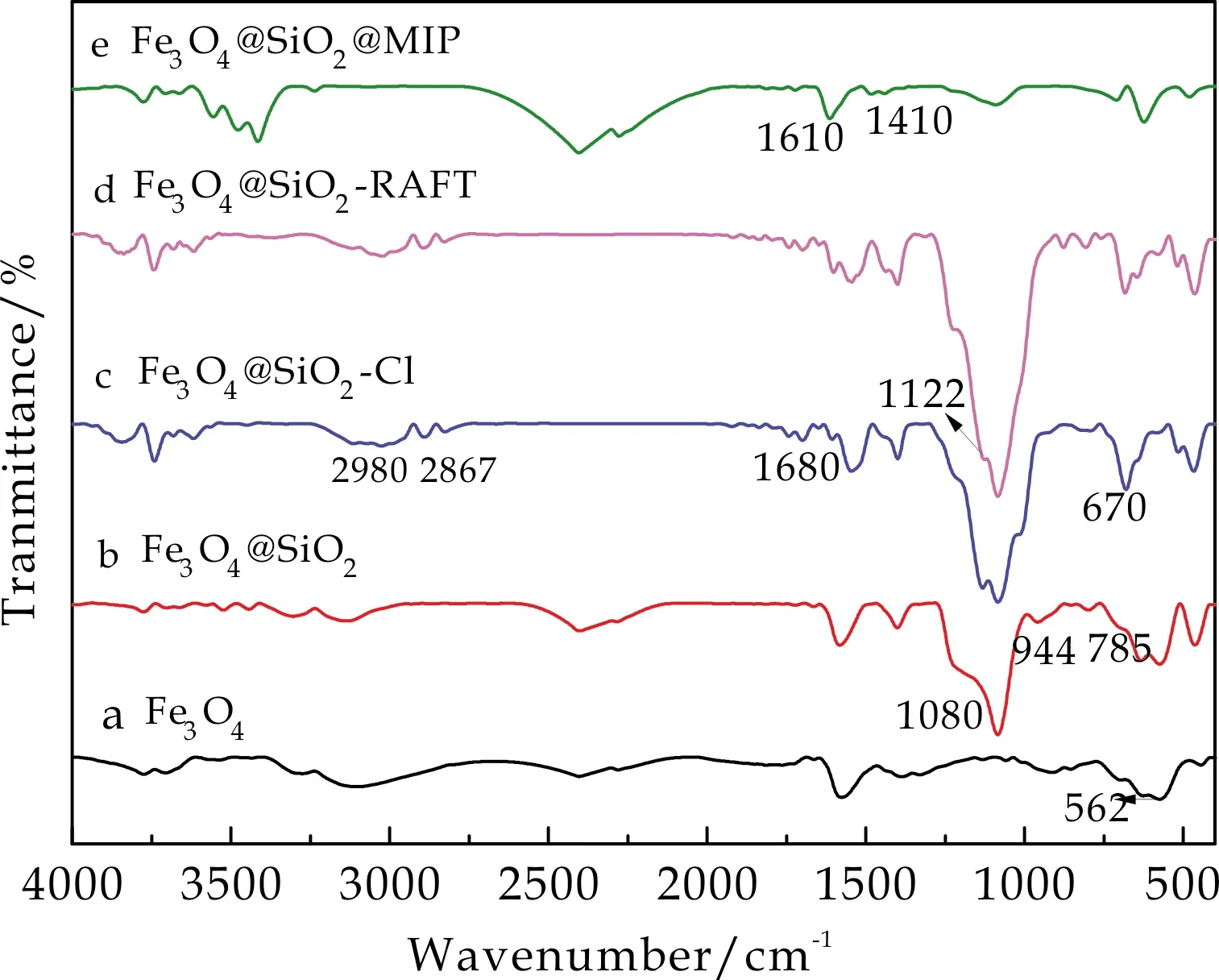
图2 各阶段产物的FTIR图
2.3 磁性能分析
通过振动样品磁强计分析了材料的磁性能变化,结果如图3所示.其中,Fe3O4纳米颗粒的比饱和磁化强度值为73.14 emu/g,而Fe3O4@SiO2和Fe3O4@mSiO2@MIP分别为36.53 emu/g和17.98 emu/g,这是因为在Fe3O4表面经过了SiO2和印迹聚合物的逐层包覆,非磁性物质占比增加导致所得磁性复合材料的比饱和磁化强度值减小.但是其仍然满足磁分离要求,可以在外加磁场的作用下对Fe3O4@mSiO2@MIP实现有效的磁分离.
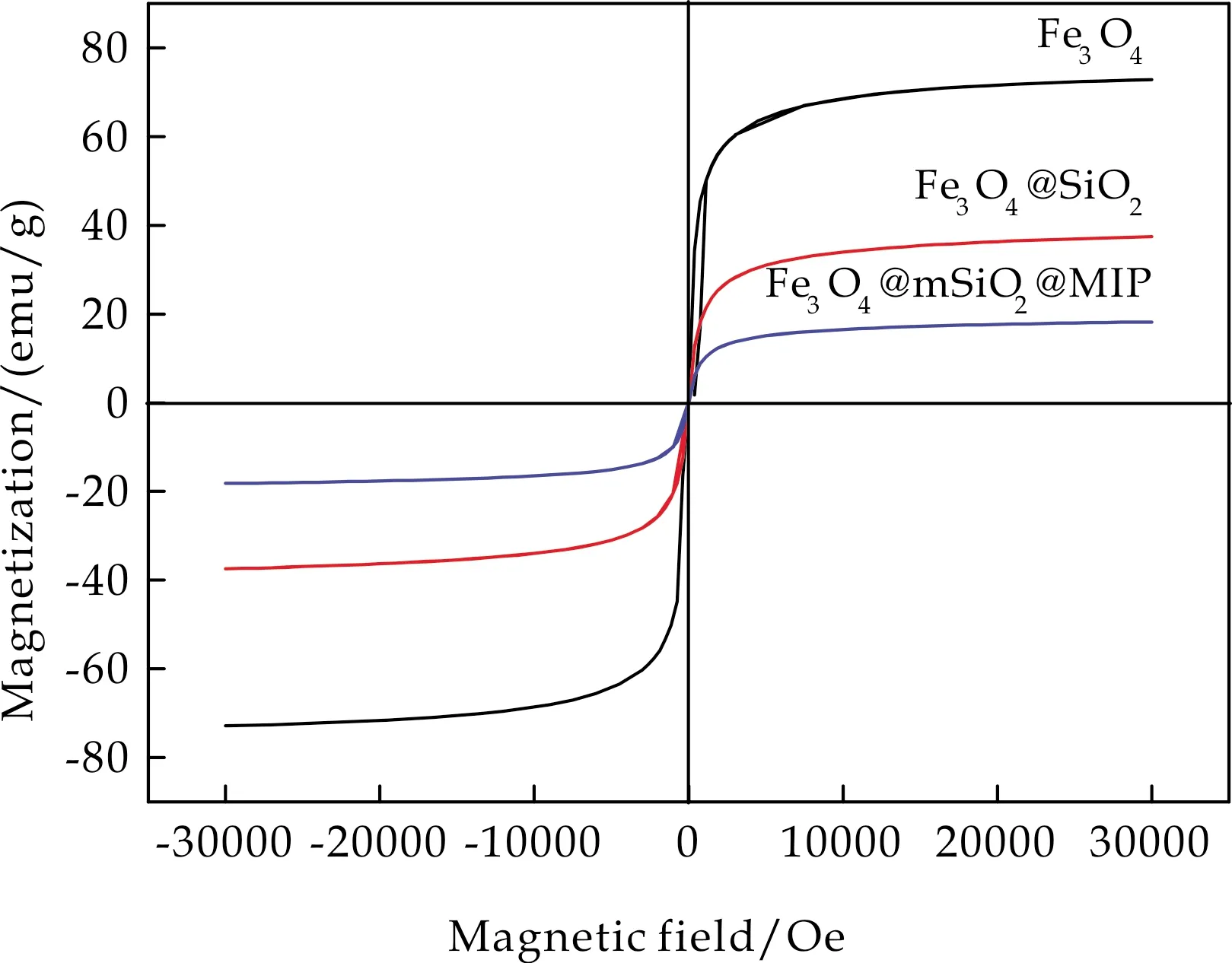
图3 Fe3O4、Fe3O4@mSiO2、Fe3O4@mSiO2@MIP的磁滞回线
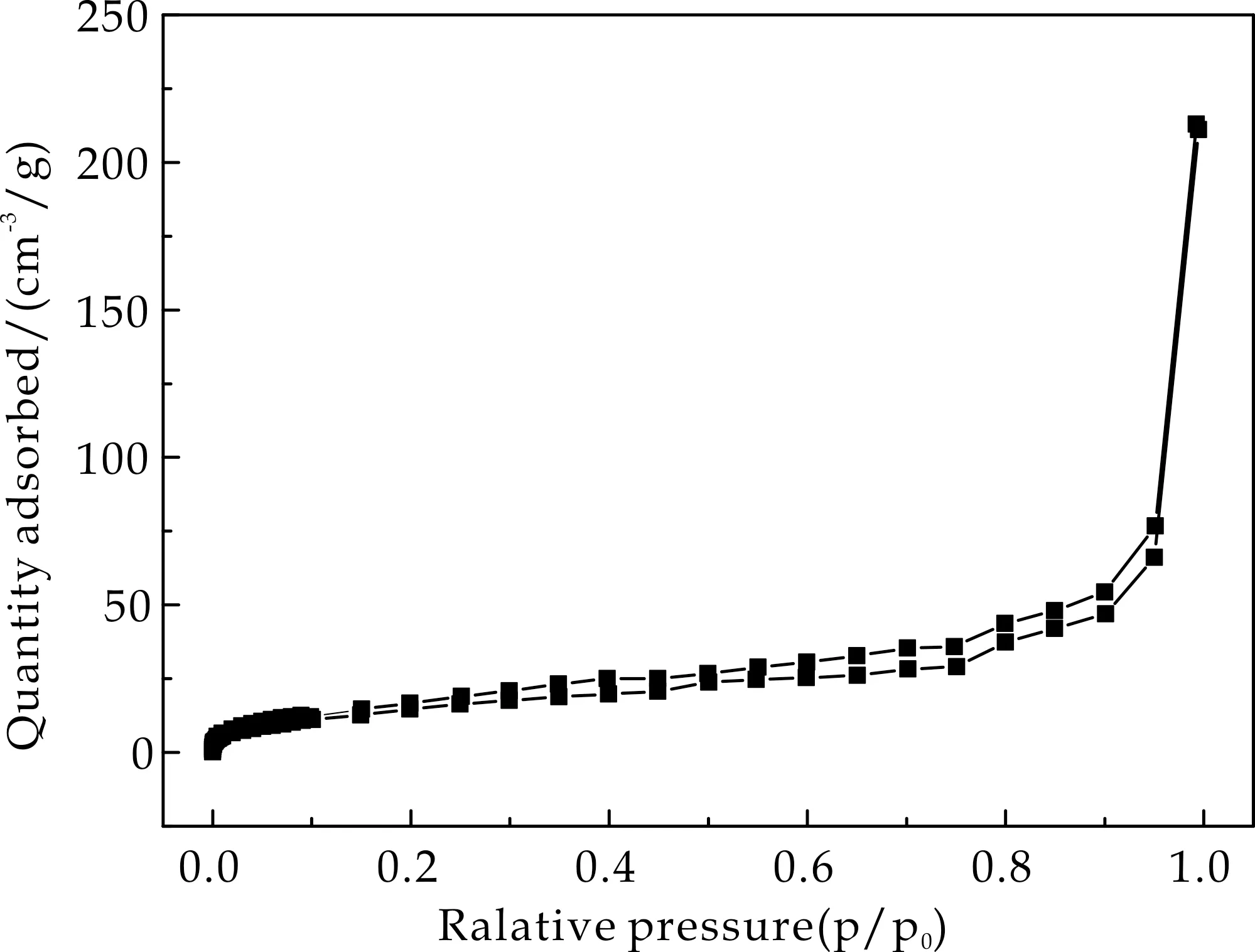
2.4 比表面积分析
通过氮气吸-脱附实验分析了Fe3O4@mSiO2、Fe3O4@SiO2以及Fe3O4@mSiO2@MIP/NIP的比表面积,结果如图4所示,其均显示出典型的Ⅳ型曲线.图4(a)和图4(b)分别为Fe3O4@mSiO2和Fe3O4@SiO2的氮气吸脱附曲线,由Brunauer-Emmett-Teller法计算得到介孔载体的比表面积为119.05 m2/g,而非介孔载体的比表面积仅为75.28 m2/g.较大的比表面积可为印迹聚合物提供更多的可识别位点,从而使其成为理想的载体.
Fe3O4@mSiO2@MIP和Fe3O4@mSiO2@NIP的氮气吸脱附曲线分别显示于图4(c)和图4(d)中,其比表面积分别为65.86 m2/g和60.29 m2/g.相比于介孔载体,比表面积的减小也说明了印迹聚合物层成功的包覆于载体表面.
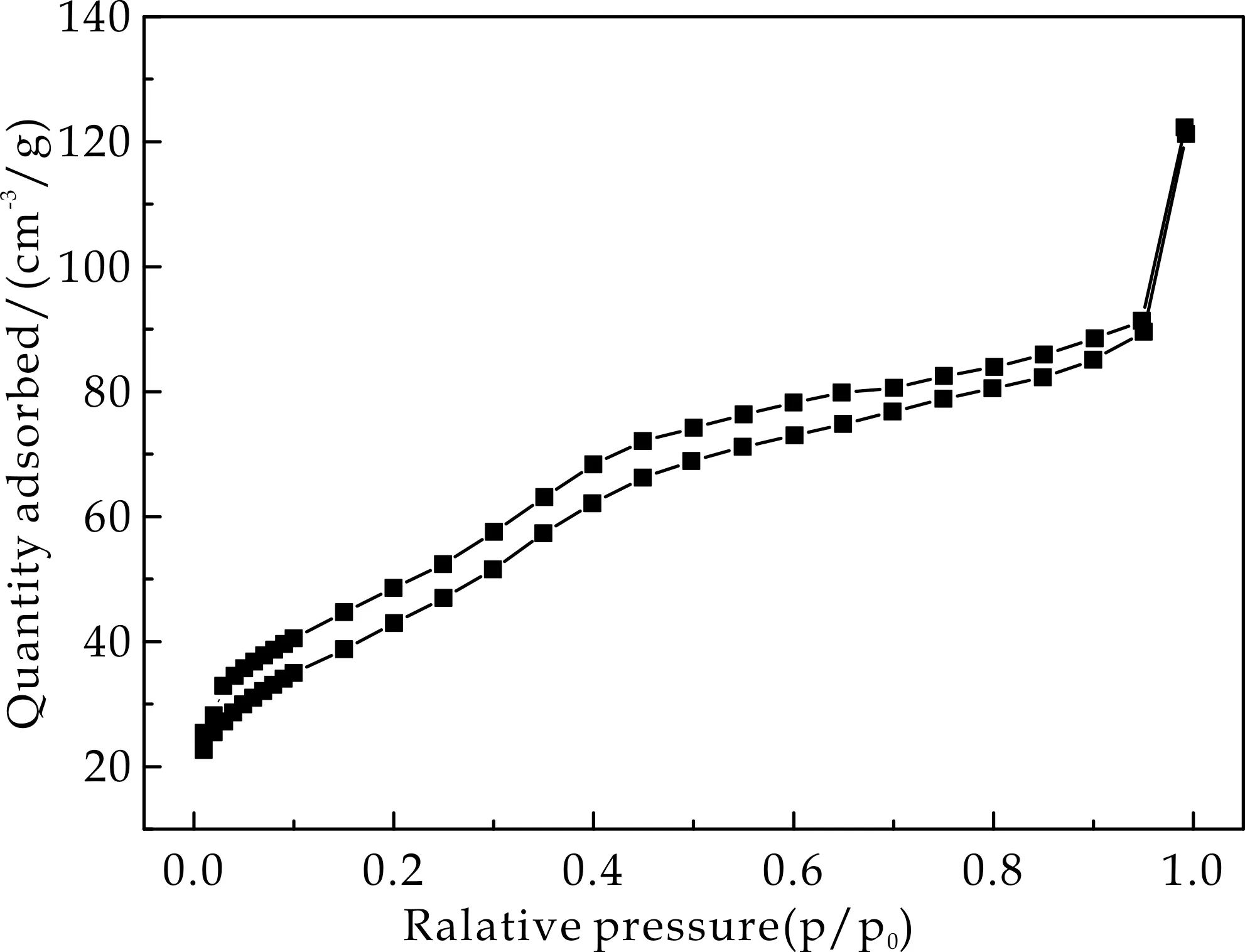
(a)Fe3O4@mSiO2
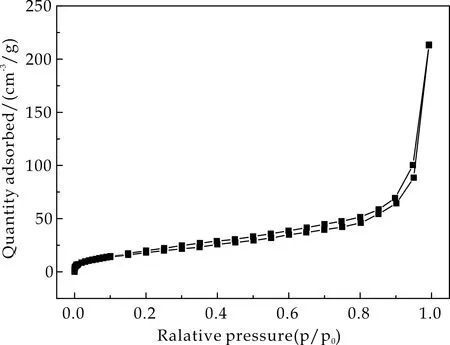
(b)Fe3O4@SiO2
(c)Fe3O4@mSiO2@MIP
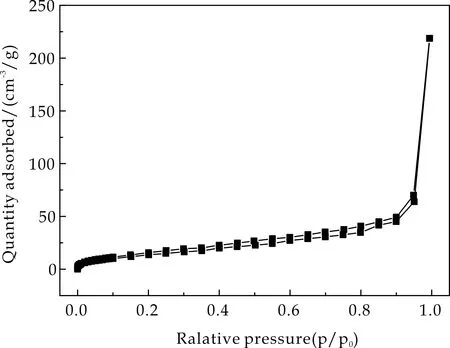
(c)Fe3O4@mSiO2@NIP图4 介孔载体、非介孔载体以及Fe3O4@mSiO2@MIP/NIP的氮气吸脱附等温线
2.5 吸附性能研究
2.5.1 吸附等温实验
图5为Fe3O4@mSiO2@MIP和Fe3O4@mSiO2@NIP对TBBPA的等温吸附曲线.从图5可以看出,Fe3O4@mSiO2@MIP和Fe3O4@mSiO2@NIP对TBBPA的吸附量均随着TBBPA浓度的增加而增加,当TBBPA的浓度达到60 mg/L后,吸附量基本达到平衡.并且Fe3O4@mSiO2@MIP对TBBPA的吸附量明显高于Fe3O4@mSiO2@NIP的吸附量,说明Fe3O4@mSiO2@MIP对TBBPA具有更高的亲和性.其原因在于所制备的印迹聚合物在去除模板分子后留下的印迹腔与模板分子很好的匹配性,而TBBPA是这个聚合过程中的模板分子.而对于Fe3O4@mSiO2@NIP,聚合层中没有与TBBPA相匹配的印迹腔,仅仅表现为非特异性吸附,因此吸附量有限.
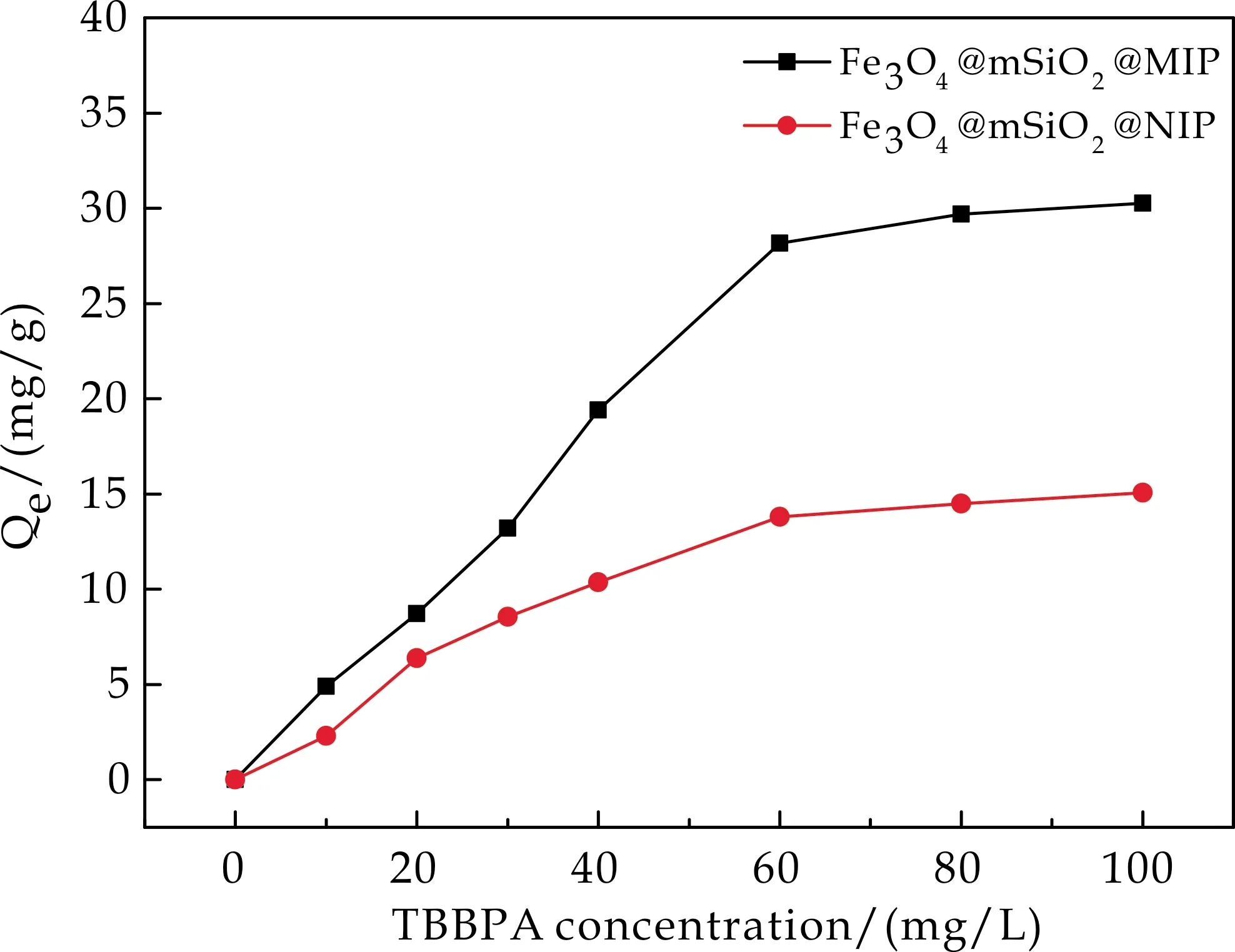
图5 Fe3O4@mSiO2@MIP/NIP的等温吸附曲线
2.5.2 吸附动力学实验
Fe3O4@mSiO2@MIP和Fe3O4@mSiO2@NIP的吸附动力学曲线如图6所示.从图6看出,随着时间的增加,Fe3O4@mSiO2@MIP对于TBBPA的吸附量先迅速增加然后逐渐趋于平衡;在20 min之后,Qt基本保持不变,曲线接近于平衡,说明达到了吸附平衡.对于Fe3O4@mSiO2@NIP来说,其对于TBBPA的吸附量随时间的变化趋势与Fe3O4@mSiO2@MIP基本一致,但其Qt值明显小于Fe3O4@mSiO2@MIP.说明印迹位点的存在使得印迹聚合物对TBBPA的结合能力优于非印迹聚合物,从而表现出更高的平衡吸附容量.
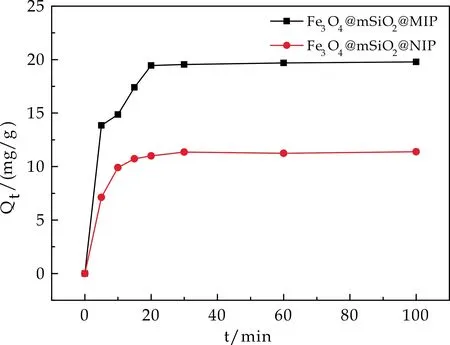
图6 Fe3O4@mSiO2@MIP/NIP的吸附动力学曲线
2.5.3 选择性吸附实验
图7显示了Fe3O4@mSiO2@MIP/NIP对TBBPA的选择性吸附性能.由图7可以看出,Fe3O4@mSiO2@MIP对于TBBPA的去除效率高达62.40%,不仅远高于对其他结构类似物的去除效率,也超出了Fe3O4@mSiO2@NIP对于TBBPA的去除效率.这是由于Fe3O4@mSiO2@MIP表面的聚合层中存在大量与TBBPA在分子尺寸、相互作用力等相匹配的识别位点,因此Fe3O4@mSiO2@MIP对TBBPA表现出较高的去除率.
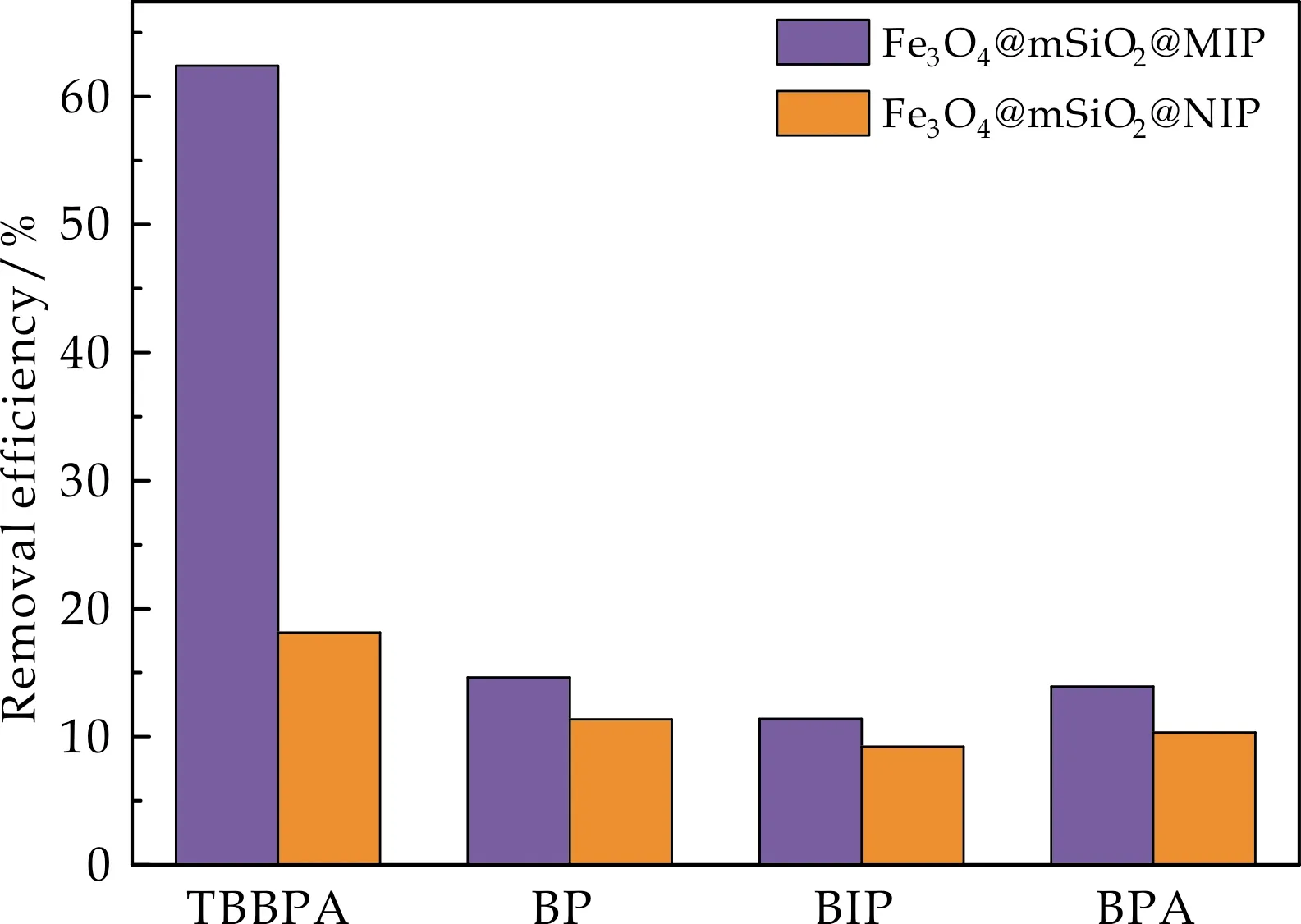
图7 Fe3O4@mSiO2@MIP/NIP对TBBPA的选择性吸附性能
2.5.4 竞争性吸附实验
采用相同浓度(50μmol/L)的TBBPA及其类似物作为待吸附物质来评价Fe3O4@mSiO2@MIP对于TBBPA特异性吸附.由图8可知,Fe3O4@mSiO2@MIP对TBBPA的去除效率明显高于对其结构类似物的去除效率,说明了印迹聚合物对于更加倾向于特异性识别TBBPA.这是由于TBBPA被作为模板分子来制备印迹聚合物,使得所形成的结合位点能够对TBBPA具有更好的识别作用.
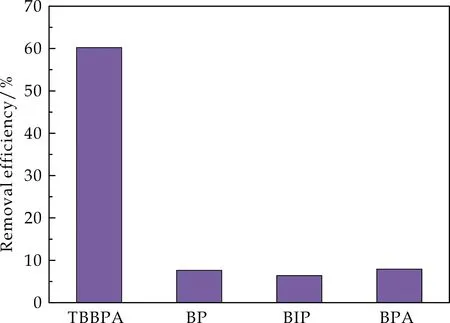
图8 Fe3O4@mSiO2@MIP的特异性吸附性能
3 结论
(1)以Fe3O4@mSiO2为载体,通过在其表面引发的RAFT聚合成功制备了分散均匀的用于快速检测TBBPA的磁性表面分子印迹聚合物Fe3O4@mSiO2@MIP纳米粒子.
(2)Fe3O4@mSiO2@MIP表现出对TBBPA优异的吸附性能,RAFT聚合和Fe3O4@mSiO2的结合使其具有更多的有效识别位点,最大吸附量为55.59 mg/g,并且可以在20 min内达到吸附平衡;选择性和竞争性测试证实了Fe3O4@mSiO2@MIP对于TBBPA的特异性吸附性能.同时,磁芯的存在有利于印迹材料的快速分离.
