近红外荧光探针用于阿尔茨海默病标志物检测的研究进展
2020-05-11葛亦然李玉艳徐云根
葛亦然,杨 剑,李玉艳,徐云根
(中国药科大学药物化学系,南京 211198)
阿尔茨海默病(AD)是一种多发于老年人的慢性神经系统退行性疾病,它由德国医生阿罗伊斯·阿尔茨海默于1906年首次公开报道。2015年,全球约有4 680万人遭受AD的折磨,且每年约有500万的新增病例,预计到2050年全球AD患病人数将达到1.15亿[1]。年龄是AD发病的最主要因素。65岁以下人群的发病率约为1%,而这一趋势随年龄增长显著上升,西方国家85岁以上的老年人口中AD的患病率约为33%[2]。AD起病隐匿,病程漫长,发病原因复杂,除年龄外,遗传因素、心血管疾病、吸烟、肥胖、糖尿病等危险因素均与AD的发病密切相关[3]。AD发病的早期症状主要为记忆障碍,随着病程的发展,患者认知功能下降,出现视空间技能损害、运动和语言障碍等症状,身体机能逐渐丧失,最终导致死亡[4]。
本文介绍了NIRF探针的研究进展,总结了多种探针的优劣并对后续研究方向进行了展望。
AD药物研发是临床药物开发失败率最高的领域之一,近年来有多个以β-淀粉样蛋白(β-amyloid,Aβ)为靶标的药物止步于Ⅱ期或Ⅲ期临床,如强生/辉瑞的bapineuzumab、罗氏公司的gantenerumab、礼来公司的solanezumab和semagacestat、默克公司的verubecestat及勃林格约翰公司的BI409306等。目前被美国食品药品安全管理局(FDA)批准用于治疗AD的上市药物有5种:盐酸多奈哌齐(donepezil hydrochloride)、氢溴酸加兰他敏(galanthamine hydrobromide)、盐酸美金刚(memantine hydrochloride)、卡巴拉汀(rivastigmine hydrogen tartrate)及他克林(tacrine)[5]。
1 AD生物标志物成像技术
尽管全球对AD的研究已有100多年,但目前仍缺乏有效的治疗手段。早期诊断技术的缺乏,使得AD治疗药物在开发时往往选取已出现明显症状的中晚期患者,此时患者脑内已积累了大量不可逆转的神经损伤,这被认为是AD药物研发高失败率的主要原因之一[6]。目前AD的临床诊断主要基于NINCDS-ADRDA定义的AD诊断标准、患者家族疾病史以及临床表现,因此诊断往往缺乏准确性[7-8]。因此,开发可用于AD早期发现和监测的诊断工具是AD药物研发的基础和当下研究的重点。
在过去的几十年里,包括计算机断层扫描(CT)、磁共振成像(MRI)、正电子发射断层扫描(PET)、单光子发射计算机断层扫描(SPECT)和光学成像在内的多种脑部成像技术得到了巨大发展[8]。MRI和PET是目前临床上使用最为广泛的成像技术。但MRI的灵敏度较低,其空间分辨率只能检测出直径大于50 μm的斑块或纤维缠结,而Aβ斑块的直径一般为20~60 μm[9]。与之相比,PET和SPECT的灵敏度较高,且能够用于观测靶点和配体之间的相互作用,其对人体内特定生物分子的检测限低至皮摩尔级别[10]。目前已有3种PET探针经FDA批准上市:[18F]FPIB(VizamylTM)、[18F]AV-45(AmyvidTM)和[18F]AV-1(NeuraceqTM),另有多个PET和SPECT探针处于临床试验阶段。但PET价格昂贵,其所用同位素含量稀缺且半衰期短(11C半衰期为20.4 min,18F半衰期为109.7 min),大大限制了PET探针的临床应用[11]。SPECT虽价格较低且所用同位素半衰期较长(125I、123I和99mTc的半衰期分别为60.1 d、13.2 h和6.02 h)[10],但存在背景干扰信号较强和血脑屏障(BBB)透过率低等不足。
与放射性核成像技术相比,光学成像因其具有无创、非辐射、价格低廉、可实时进行体内外多靶点监测等优势而受到广泛关注。光学成像最大的不足是难以达到检测所需的穿透深度,而波长范围在650~900 nm的近红外光具有较好的穿透深度,能够适应体内检测需求,且该波长范围下生物物质的自荧光干扰最小,对生物样品的光损伤也最小[12]。目前已有多个近红外荧光染料被开发用于体内检测,如花菁染料(Cy7)、Alexa荧光染料、SRfluor染料等[13]。其中吲哚菁绿(ICG)和亚甲蓝(MB)经FDA批准用于临床。目前近红外荧光探针主要应用于肿瘤成像领域,开发可用于检测神经退行性疾病的NIRF探针具有极大的研究意义和挑战性。
2 用于检测Aβ的NIRF探针
“淀粉样蛋白级联”假说由John Hardy和Dennis Selkoe于1991年首次提出,是最为广泛认可的AD假说之一。该假说认为,AD病程始于脑内Aβ生成与清除的失衡,Aβ的逐渐聚集和沉积最终导致了AD的发病[14]。Aβ单体由淀粉样前体蛋白(APP)水解产生。APP在β-和γ-分泌酶作用下生成Aβ40和Aβ42单体,然后经过构象变化形成富含β-折叠的寡聚体形式,再经过进一步聚集沉积最终形成不可溶的Aβ纤维和斑块[5]。近期有研究表明,在不同Aβ聚合状态中,可溶性Aβ寡聚体的神经毒性最强[15-16]。尽管自“淀粉样蛋白级联”假说提出的近30年来,人们提出了多项证据以验证该假说,但AD发病的具体生理变化和分子机制尚不明确。
2.1 NIAD-4及其类似物
NIAD-4是最早报道用于Aβ斑块体内成像的NIRF探针之一,它于2005年由Nesterov等报道。该化合物采用了经典的推-拉电子结构,其中对羟基苯基作为电子供体,双氰基甲基作为电子受体,二者由二噻吩共轭链桥连。NIAD-4与Aβ聚合物作用后荧光强度升高约400倍,与Aβ的亲和力强(Ki=10 nmol/L)。体内双光子实验表明,NIAD-4静脉注射后能够有效透过转基因小鼠的BBB并标记脑内及血管中的Aβ斑块(图1)。但是,该探针的最大发射波长仅有612 nm,未能达到近红外窗。
图1 (A)NIAD-4的脑切片染色实验[17];(B)体内双光子荧光成像实验[17]
为了获得波长更长的化合物,该课题组随后又报道了一系列NIAD-4类似物[18]。其中NIAD-16的发射波长达到720 nm,且在不同的组织基质中显示出不同的荧光寿命分布,故可用于区分背景信号下的血管和非血管性Aβ斑块(图2)。
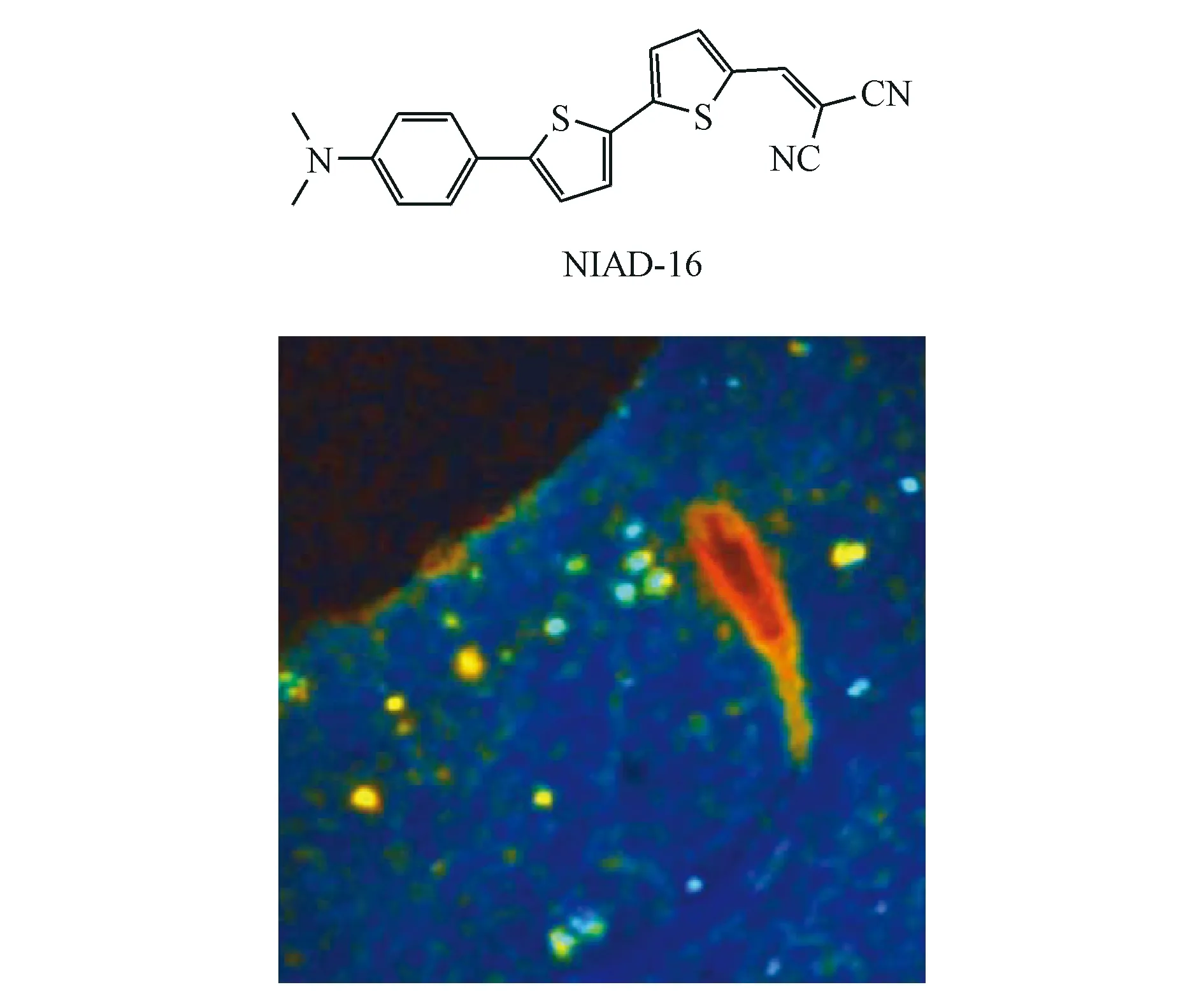
图2 NIAD-16的荧光成像实验[18]
2.2 AOI-987
同年,Hintersteiner等[19]报道了一系列用于Aβ成像的嗪类染料。其中AOI-987的激发/发射波长达到650/670 nm,对Aβ聚合物结合能力较好(Kd=220 nmol/L)。脑切片染色实验显示,AOI-987能够选择性标记Aβ斑块(图3-A)。体内试验结果表明,AOI-987能够有效穿透BBB并标记脑内Aβ斑块(图3-B)。该化合物最大的缺陷是脑内清除速率较低。
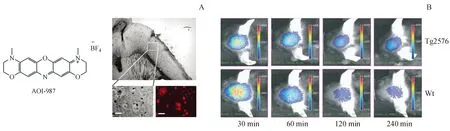
图3 (A)AOI-987的脑切片染色实验;(B)AOI-987的体内试验[19]
2.3 CRANAD-X
2009年,Ran等[20]报道了首个姜黄素类似物NIRF探针CRANAD-2。姜黄素具有广泛的抗肿瘤、抗氧化和抗炎等活性,且被用作抗淀粉样蛋白物质[21]。与Aβ聚合物孵育后,CRANAD-2的荧光强度升高约70倍,波长蓝移达到90 nm,且其与Aβ聚合物的亲和力达到Kd=38.0 nmol/L。体内实验中,该探针能够通过荧光强度变化迅速区分Tg2576小鼠和Wt小鼠(图4)。
该课题组随后在CRANAD-2的基础上进行了结构改造,期望获得能够标记可溶性Aβ(sAβ)的NIRF探针。于2011年报道的CRANAD-3[22]不仅能够结合Aβ聚合物,同时能够与可溶性Aβ单体和二聚体作用。体内外实验中,通过光谱分离技术可以将背景吸收、Aβ-CRANAD-3复合物和游离探针明显区分(图5-A)。
2013年,该课题组报道了更多可用于同时标记可溶性和非可溶性Aβ的小分子探针。其中CRANAD-58[23]具有良好的荧光性质(与Aβ40和Aβ42结合后荧光强度分别上升91.9和113.6倍)和Aβ单体亲和力(与Aβ40的Kd=105.8 nmol/L,与Aβ42的Kd=45.8 nmol/L),与二聚体结合的荧光强度变化与单体相近(图5-B)。
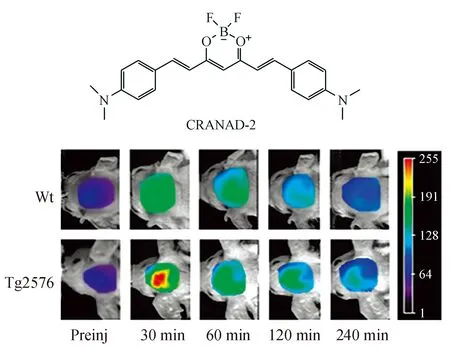
图4 CRANAD-2的体内实验[20]
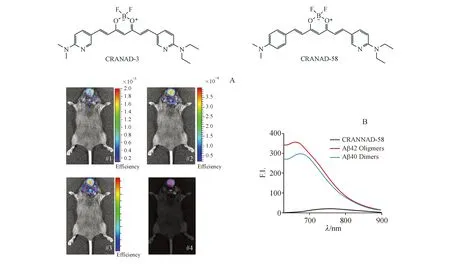
图5 (A)自发荧光非混合分布图像(#1),Aβ-CRANAD-3复合物(#2),游离的CRANAD-3(#3),复合图像(#4)[23];(B)CRANAD-58与Aβ40二聚体和寡聚体作用的荧光图像[23]
该课题组研究发现,通过调整姜黄素骨架4-位苯氧基烷基链的立体位阻可提高探针分子对sAβ的选择性[24],并于2017年报道了一系列新的化合物。其中CRANAD-102对可溶性Aβ的亲和力比不可溶性Aβ(insAβ)高68倍(Kd=7.510 nmol/LvsKd=505.9±275.9 nmol/L)(图6-A)。体内实验表明(图6-B),CRANAD-102能够检测出4个月大的APP/PS1小鼠(该年龄下的小鼠脑内以sAβ为主)。此外,CRANAD-102还能够检测出4~12个月小鼠脑内sAβ的含量变化,证明其可作为AD症状前阶段sAβ检测的一个候选化合物。
2.4 BODIPY7及其类似物
2010年,Ono等[25]报道了一系列二吡咯甲烷硼(BODIPY)类似物SPECT/荧光双功能分子探针。其中BODIPY7的激发/发射波长为606/613 nm且对Aβ聚合物表现出较好的结合能力。但BODIPY7的大脑摄取率极低,给药后探针分子大部分被肝脏快速吸收。
2012年,该课题组又报道了另一个BODIPY类似物分子探针BAP-1[26](图7-A)。该化合物有较高的量子效率(46.8%)和对Aβ聚合物较强的结合能力(Kd=44.1 nmol/L),但其激发和发射波长较短,分别只有614和648 nm。与BODIPY7相比BAP-1大大改善了脑摄取率,且在脑内的清洗速率快。脑切片染色实验结果显示BAP-1能够特异性标记AD脑内的Aβ斑块(图7-A,B)。
2013年,该课题组又报道了一系列BODIPY类似物探针BAP-2~BAP-5[27]。
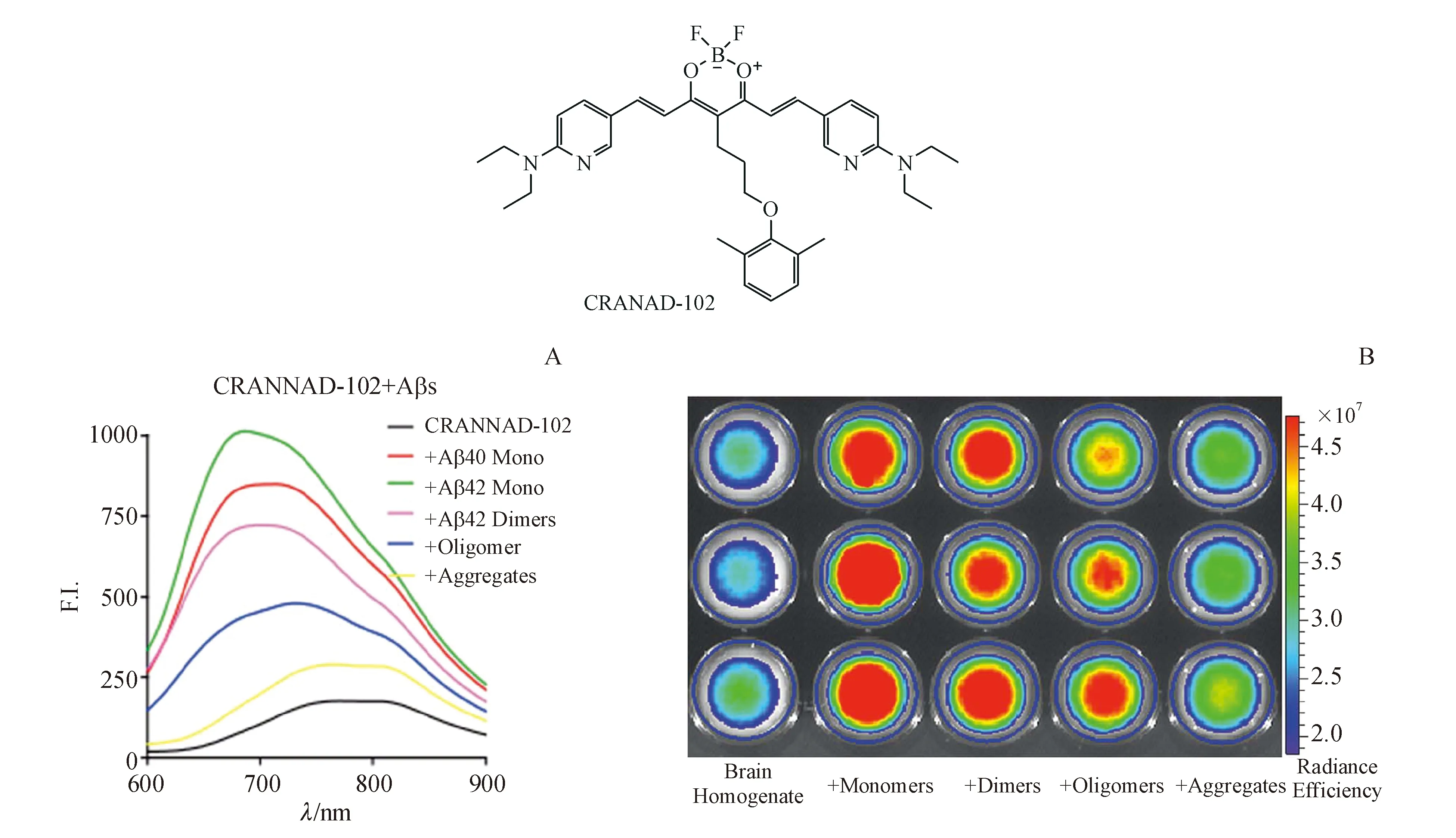
图6 (A)CRANAD-102与sAβ和insAβ作用的荧光图像;(B)脑匀浆实验[24]

图7 (A)Tg2576小鼠的脑切片染色实验;(B)Wt小鼠的脑切片染色实验[26]
2.5 THK-265
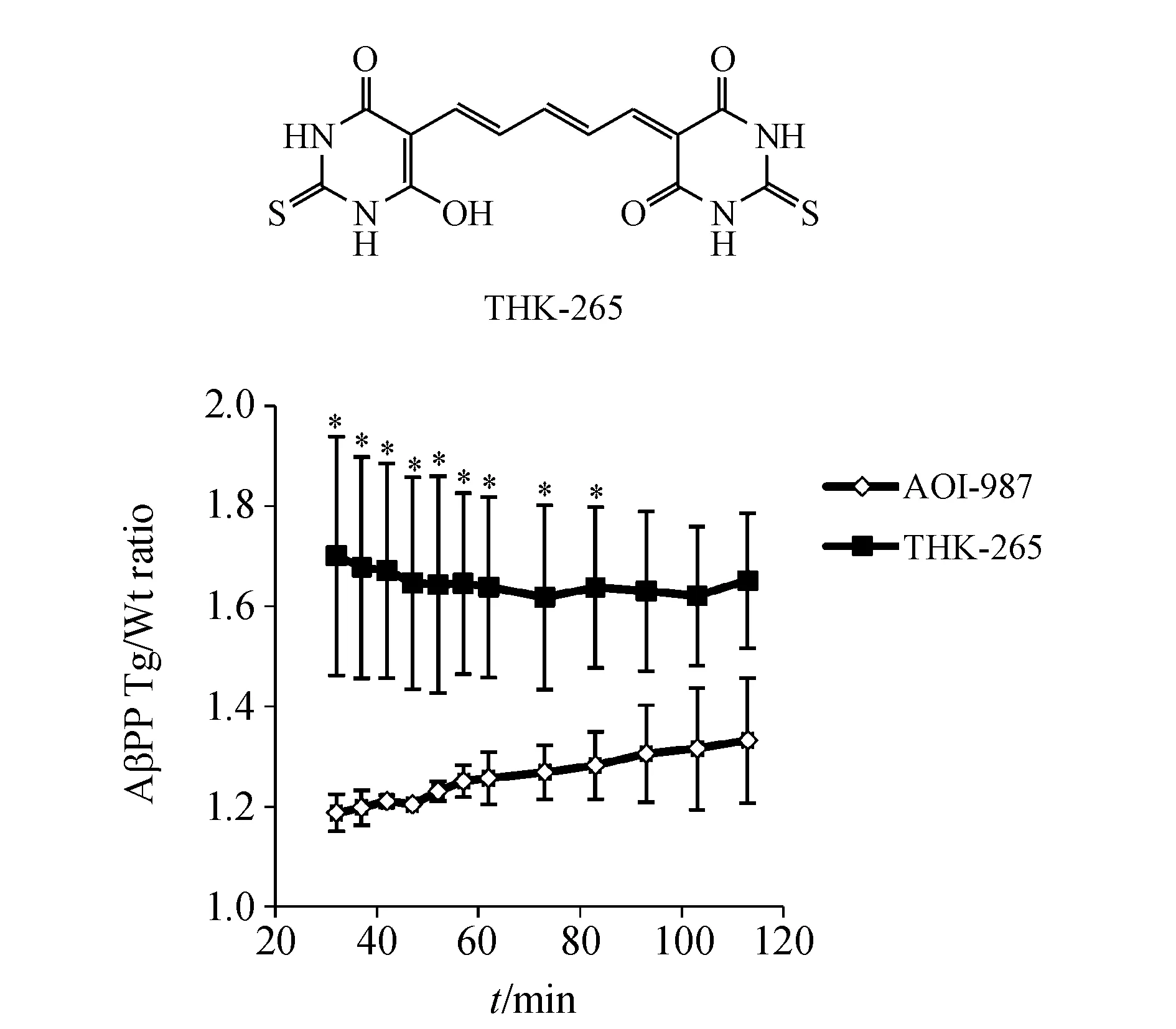
2010年,Okamura等[28]报道了THK-265作为体内检测脑内Aβ斑块的候选探针分子。THK-265有较高的量子效率(38.5%,甲醇溶液)和摩尔吸收系数(96 198 mol-1·cm-1,甲醇溶液)。其对Aβ纤维的Kd=97 nmol/L且作用后荧光强度升高6倍。值得注意的是,脑切片实验显示,THK-265主要与脑中的核心斑块结合,而对游离斑块作用较弱,这使得其检测结果受正常衰老过程中Aβ病理变化的影响较小。Schmidt等[29]随后证实了THK-265用于直接监测和评估AD动物模型中不同病理阶段大脑皮层中的Aβ水平的能力。此外,静脉注射THK-265后,其AβPP Tg小鼠与Wt小鼠的荧光强度比值(AβPP Tg/Wt)持续呈现高水平,且在30~90 min内显著高于AOI-987的比值(图8),表明其优良的成像对比度。
*P<0.05
2.6 DANIR 2c及其类似物
2013年,Cui等[30]报道了一系列新型NIRF探针NADIRs。其中DANIR 2c的发射波长为665 nm,对Aβ42聚合物有很强的亲和力(Ki=37 nmol/L,Kd=27 nmol/L)。体内实验表明,静脉给药30 min后,22个月大的Tg 2576小鼠脑部荧光强度显著高于Wt小鼠(图9)。
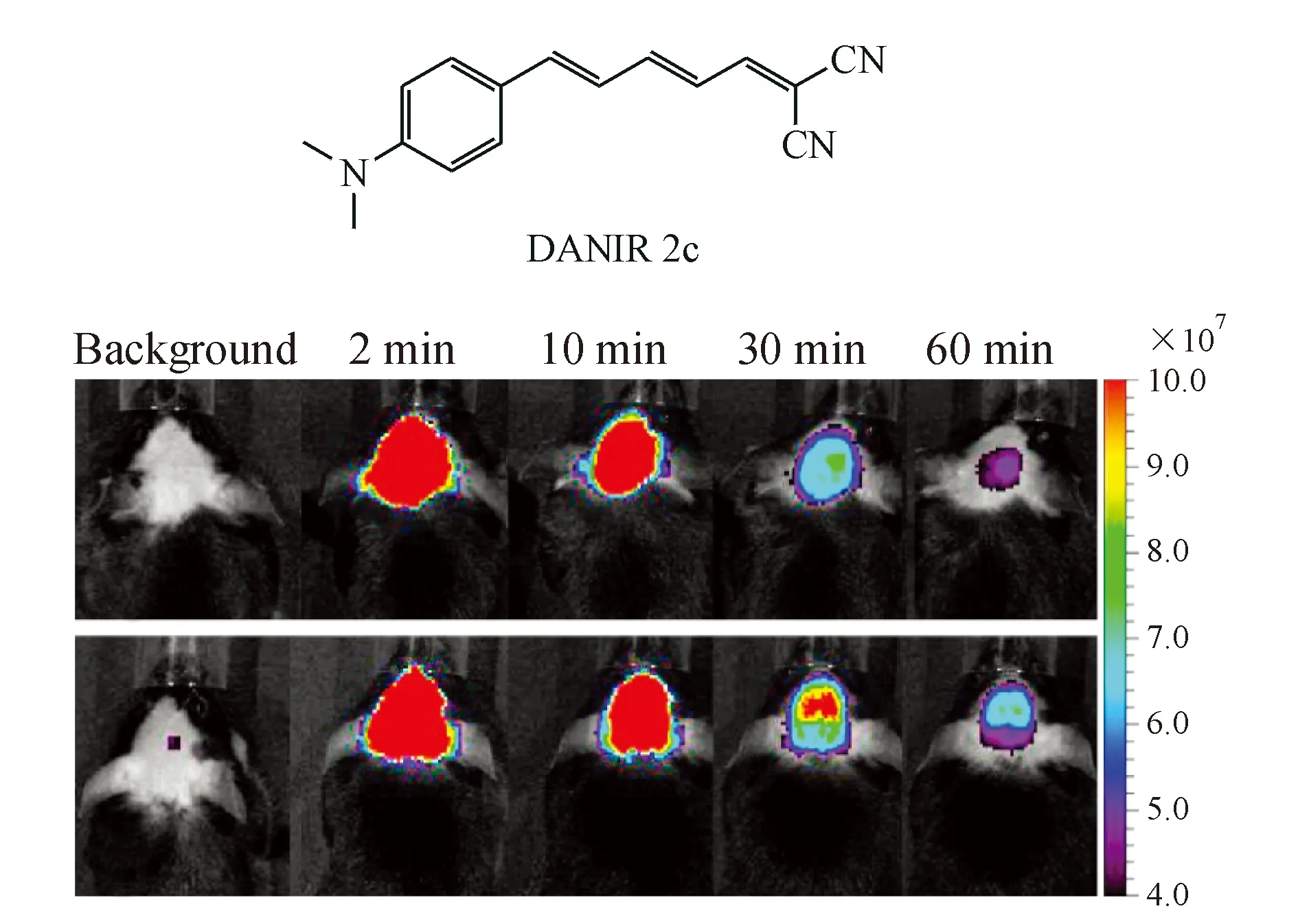
图9 DANIR 2c的体内实验[30]
为了增加化合物的发射波长,该课题组又设计合成了4个同系列探针MAAD-3、DMDAD-3、MCAAD-3和DMMAD-3[31]。脑切片染色实验证明化合物均能选择性标记Aβ斑块(图10)。
2015年,该课题组又报道了一系列同类型探针分子[32]。其中化合物DANIR 8c的发射波长达到798 nm,与Aβ聚合物作用后蓝移至678 nm,对Aβ亲和力高(Kd=14.5 nmol/L),且在脑内有着高摄取率、高清除速率和较好的稳定性。体内实验表明DANIR 8c能够有效标记脑内Aβ斑块,并区分AD转基因小鼠和Wt小鼠(图11-A)。同年,该课题组报道了化合物DANIR 3b和DANIR 3c[33],均具有较好的荧光性质、高量子效率、高BBB透过率和强Aβ亲和力(DANIR 3b:Kd=8.8 nmol/L;DANIR 3c:Kd=1.9 nmol/L),能够选择性标记Aβ聚合物(图11-B,C)。
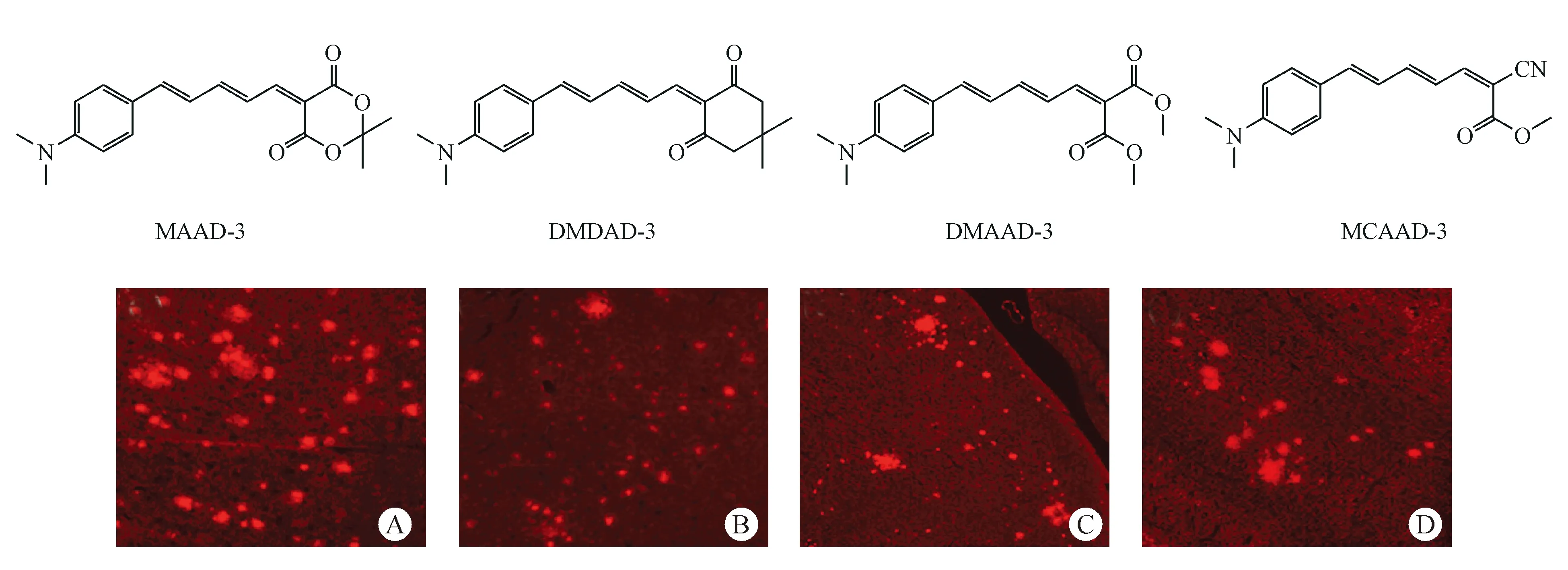
图10 脑切片染色实验[31]:(A) MAAD-3;(B) DMDAD-3;(C) MCAAD-3;(D) DMMAD-3
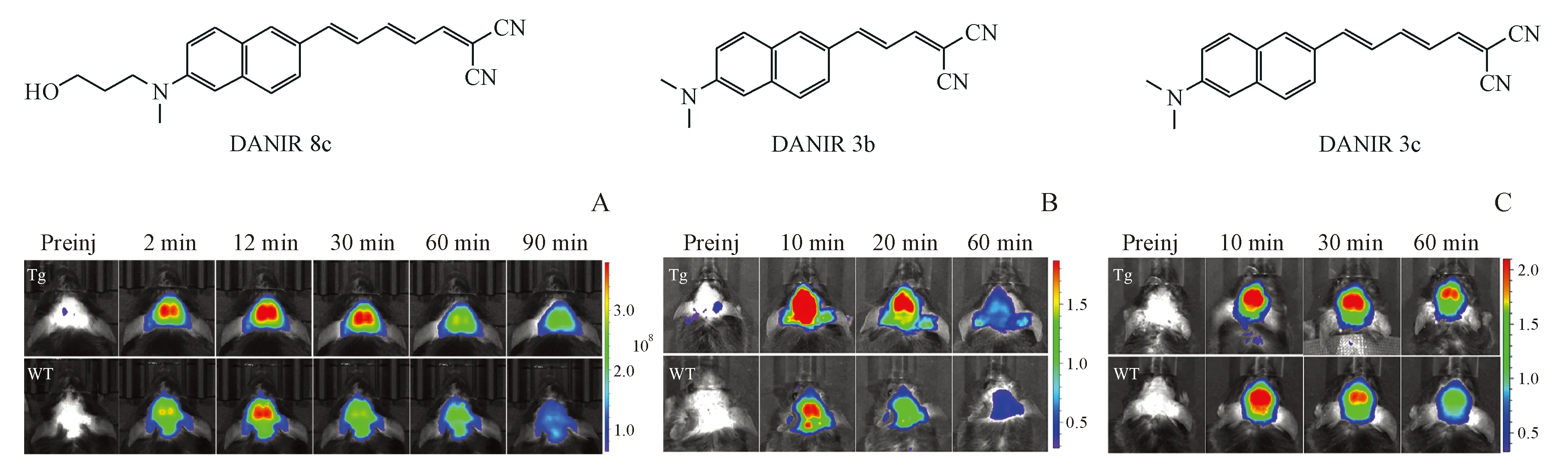
图11 (A) DANIR 8c的体内实验[32];(B) DANIR 3b和(C) DANIR 3c的体内实验[33]
2.7 纳米探针
金纳米棒(AuNR)因其良好的生物相容性和光学韧性而受到广泛研究和应用[34-35]。2017年,Qu等[36]报道了一种新型肽共轭金纳米棒(AuP)作为多功能Aβ纤维检测剂和纤维化抑制剂。AuP是AuNR和两种抑制Aβ抑制剂(Aβ15-20和POM)的复合物(图12-A,B),它能够有效抑制Aβ聚合,离解沉积物并保护神经细胞。AuP是首个集Aβ-靶向部分、信号报告部分和抑制剂于一体的探针系统,为兼具检测和治疗作用的多功能纳米材料的设计提供了方向。
2018年,Li等[37]报道了一种新型NIR/MRI双功能探针Fe3O4@SiO2@SLCONHRNPs(图13)。该探针将Aβ靶向的咔唑基花菁类染料和超顺磁性氧化铁纳米粒(Fe3O4NPs)结合,得到了无毒性、高BBB透过率的纳米探针。该探针对Aβ诱导的毒性有很强的神经保护作用,在体内实验中对标记Aβ有较高的灵敏度和选择性,且具备高MRI空间分辨率。
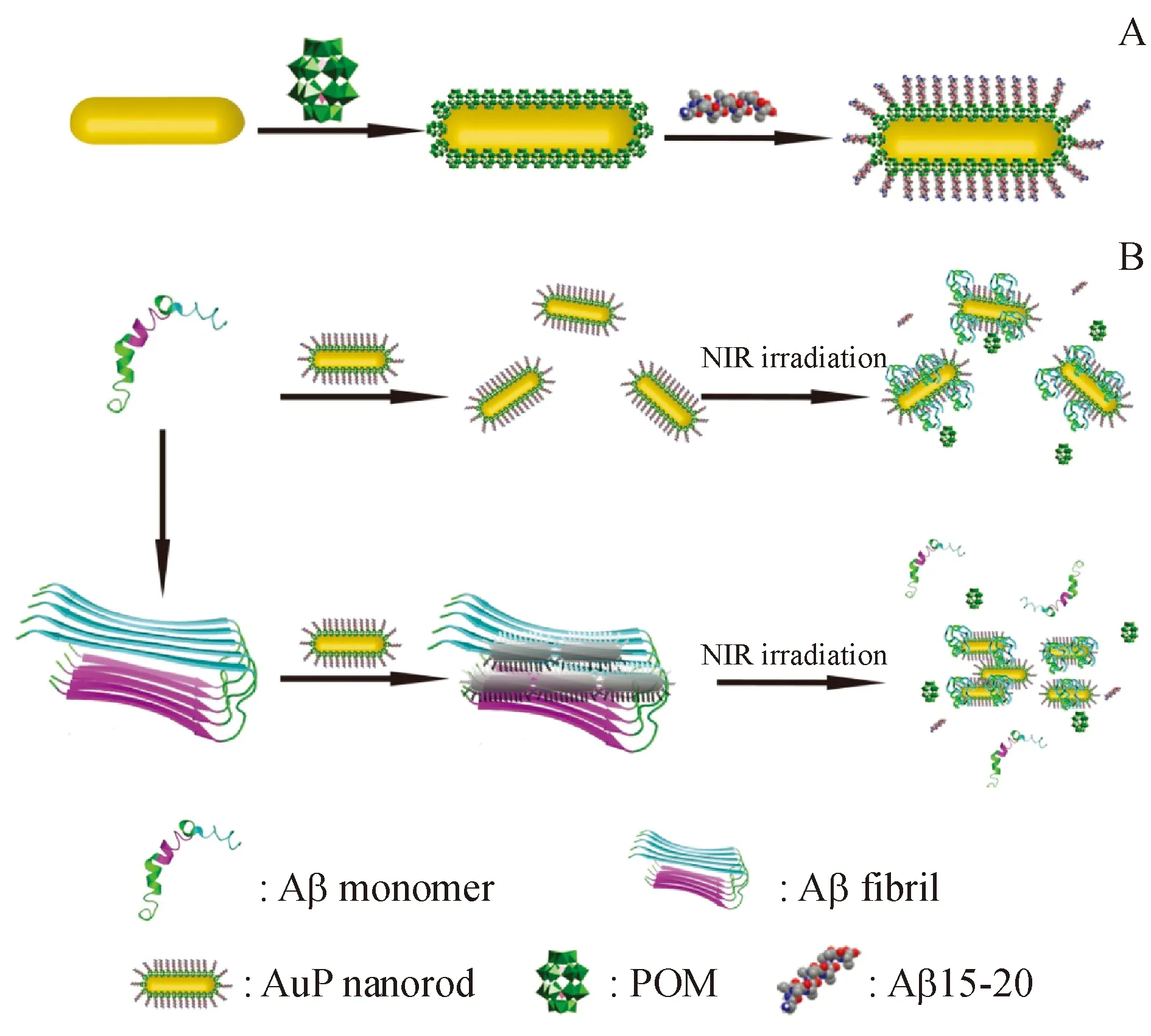
图12 (A) AuP制备示意图;(B)AuP用于AD诊断和治疗的示意图[36]
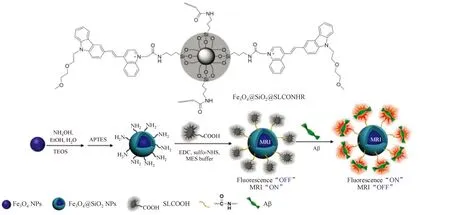
图13 Fe3O4@SiO2@SLCONHR NPs的制备示意图作用的信号响应示意图[37]
2.8 其他用于Aβ成像的NIRF探针
2016年,Li等[38]设计合成了一系列带电荷的咔唑基花菁类探针,其中化合物DBA-SLOH的吸收/发射波长为560/670 nm。亲脂性烷基链的引入使该探针具有的BBB透过率和生物相容性。DBA-SLOH在脑内显示出对Aβ良好的特异性,但其与Aβ的亲和力并不理想(Aβ40纤维:Kd=1.13 μmol/L,Aβ40单体:Kd=6.18 μmol/L)。
2017年,Pascal等[39]报道了一系列能够标记Aβ斑块并阻止Aβ聚合的化合物,其中化合物5a1的发射波长达到700 nm,并显示出极强的Aβ亲和力(Kd=10.2 ± 1.1 nmol/L)。脑切片实验证实其对Aβ斑块的选择性标记能力。化合物5a1对Aβ聚合的抑制作用(IC50=1.8~14.6 μmol/L)强于姜黄素(IC50=17.4 μmol/L)。
同年,具有双光子吸收和近红外波长的化合物DCIP-1被报道用于Aβ斑块成像[40]。体内实验验证其BBB透过能力和活体标记AD小鼠脑内Aβ斑块的能力。
同年,Rajasekhar等[41]报道了香豆素喹啉共轭类NIRF探针CQ用于特异性检测Aβ聚合物。与Aβ聚合物作用后,CQ的荧光强度升高100倍,结合能力Ki=86 nmol/L。此外,该化合物对Aβ聚合物有较强的特异性,能有效区分Aβ和Tau蛋白、α-突触核蛋白(α-Syn)及胰岛淀粉样多肽(IAPP)。
2018年,Lee等[42]报道了一种带电荷的NIRF探针Kim 1。与Aβ聚合物孵育后,该化合物在710 nm处出现显著的荧光强度增强,且荧光响应值随Aβ聚合程度而变化。
3 用于检测Tau蛋白的NIRF探针
神经纤维缠结(NFT)是AD的另一个重要的病理标志物,它由异常磷酸化的Tau蛋白组成。Tau蛋白是一种正常的轴突蛋白,它通过与微管结合,促进微管的组装和稳定[43]。研究表明,AD脑中Tau蛋白的磷酸化程度比正常大脑高出3~4倍。过度磷酸化的Tau蛋白可导致微管解体,影响轴突运输,从而损害神经元和突触功能[43]。过度磷酸化的Tau蛋白还可聚集成成对螺旋细丝(PHF),进而在神经元中聚集积累形成NFT[44-45]。
3.1 CyDPA2
2013年,Shao等[46]报道了首个用于Tau蛋白检测的NIRF探针CyDPA2。该探针将吲哚菁绿和二烟酰胺锌(DPA-Zn)共轭连接,从而与Tau聚合物的磷酸化位点结合。CyDPA2的吸收/发射波长为826/840 nm,对磷酸化Tau蛋白(pTau)的亲和力强(EC50=0.304 μg/mL)。在比率测量中,CyDPA2 与pTau和非磷酸化Tau蛋白(nTau)作用后,比率吸收分别增加17%和减少25%(图14-A)。离体脑提取物实验中,探针与晚期AD患者脑提取物和P301L小鼠脑提取物孵育后吸收比分别增强13%和10%。在凝胶染色实验中,CyDPA2可明显标记AD脑、P301L小鼠脑和pTau样品的磷酸化蛋白带,而对nTau对照样品无明显作用(图14-B)。荧光显色实验进一步验证了上述结果(图14-C)。值得一提的是,CyDPA2能够对Tau蛋白病理学进行视网膜成像,从而有效避免透光深度不足问题。
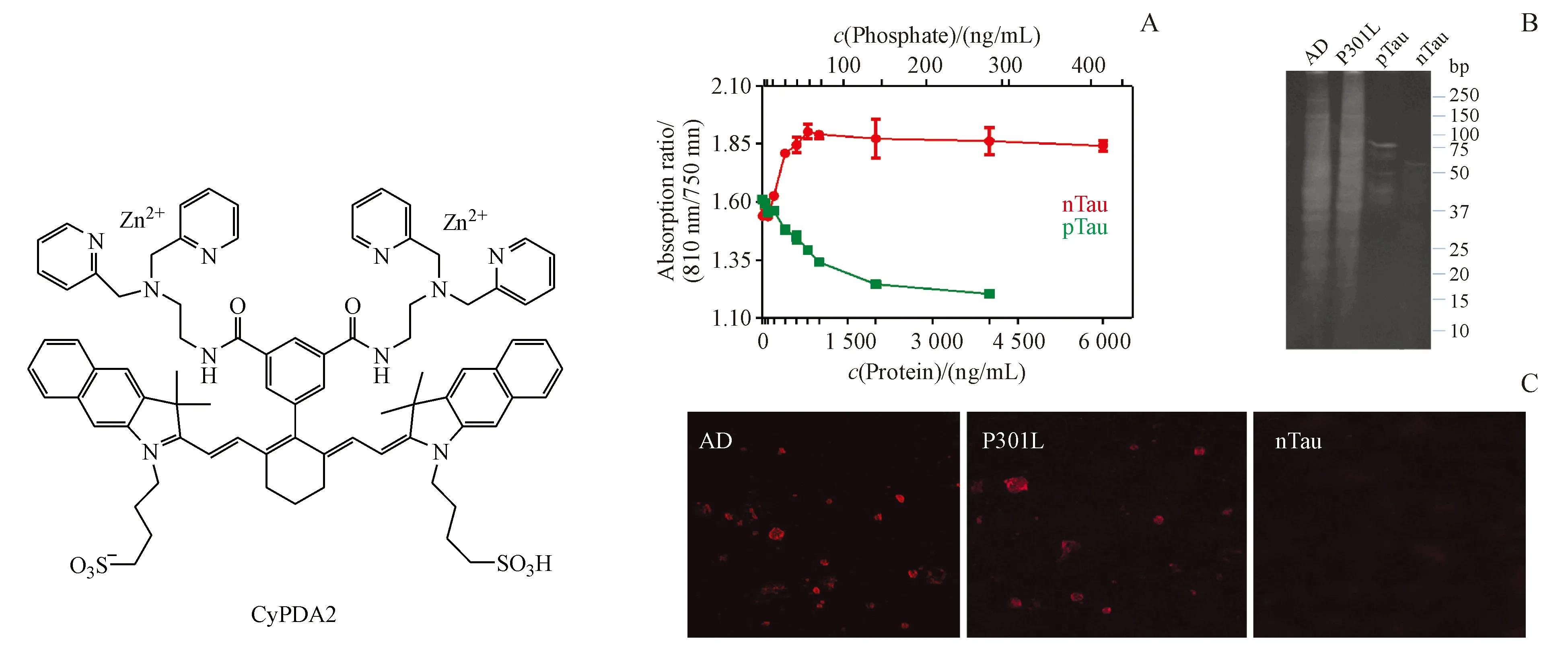
图14 (A)CyDPA2与pTau和nTau作用的吸收率变化图;(B) CyDPA2的凝胶染色实验;(C)CyDPA2的荧光显微镜成像实验[46]
3.2 化合物 1c
2015年,Park等[47]报道了一系列以姜黄素为骨架的Tau纤维探针。其中化合物1c的发射波长为620 nm,与Tau蛋白作用后量子效率增加22.9倍,与Tau蛋白的Kd=0.77 μmol/L。细胞显色实验表明,化合物1c能够检测出活细胞(Tau-GFP转染的SHSY-5Y细胞株)中的Tau聚合物。但该化合物与Tau聚合物结合后波长几乎无变化,且其对Tau蛋白选择性较差,对Aβ也有显色标记作用。
3.3 Tau 1和Tau 2
2017年,Peter等[48]报道了两个BODIPY类似物Tau选择性探针Tau 1和Tau 2。为了获得较高的Tau选择性,该化合物的设计主要基于以下两条理论:(1)当化合物分子中电子供体和受体间的距离在13~19 Å时,对Tau蛋白有较好的选择性[49]。(2)在分子中引入稠环系统可提高Tau选择性[48]。探针Tau 1和Tau 2均显示出良好的光学性质,发射波长分别达到705和687 nm(氯仿溶液),且有较大的斯托克斯位移。与Tau蛋白和聚阴离子肝素(诱导Tau蛋白聚集)孵育后,化合物的荧光强度呈现明显的时间依赖性增强,孵育48 h后分别升高6.4和9.3倍(图15-A,B),且化合物与Aβ孵育后荧光几乎无变化(图15-C,D),证明Tau 1和Tau 2对Tau蛋白良好的选择性。脑切片染色实验显示Tau 1能够选择性将脑组织中的Tau聚合物染色(图15-E),体内实验结果表明Tau 1能够有效穿透BBB并通过标记区分转基因小鼠、Wt小鼠和阴性对照(图15-F)。共聚焦成像实验的结果进一步验证了以上结论(图15-G)。
4 用于检测ROS的NIRF探针
多项证据表明,氧化应激在AD的病理机制中起关键作用[50]。在正常生理条件下,人体内的活性氧簇(reactive oxygen species,ROS)含量维持在低水平,年龄和疾病导致的线粒体功能丧失、金属离子稳态失衡、抗氧化能力缺失等原因可导致ROS生成增加或清除减少,进而引起氧化应激反应,影响突触活动和神经递质传递[51-52]。ROS还可直接攻击脑细胞内的线粒体、核酸、脂质和蛋白,破坏细胞内稳态,导致细胞凋亡。证据表明,AD大脑海马和皮层中氧化产物水平上升与Aβ40和Aβ42增加息息相关[53]。
2016年,Yang等[54]报道了首个可用于级联放大检测AD脑内H2O2水平的NIRF探针CRANAD-88。由于其结构中氨基甲酸酯的吸电子效应,CRANAD-88在近红外区是“不可见的”(激发/发射波长为580/690 nm),与H2O2作用后,硼酸酯和氨基甲酸酯部分掉落,化合物波长发生显著红移(激发/发射波长红移50/40 nm),使其在近红外区变得“可见”(图16-A)。此外,与H2O2孵育后,该探针的荧光强度升高4倍,而与H2O2和Aβ共同孵育后,荧光强度升高16倍(图16-B),呈现级联放大效应。CRANAD-88对H2O2的作用迅速而显著,对H2O2的灵敏度高于其他ROS(NO·、O2-和ClO-)。体内实验表明,APP/PS1小鼠组的荧光信号强度明显高于Wt小鼠组(图16-C)。CRANAD-88还可用于监测H2O2清除剂使用前后脑内H2O2水平(图16-D)。
2017年,该课题组报道了一个引入草酸酯基团的NIRF探针CRANAD-61用于检测AD脑内的ROS水平[55]。与H2O2孵育后,该化合物显示出显著的波长蓝移(从810 nm蓝移至570 nm),这一蓝移可用于双色双光子成像和转化型近红外成像。液质联用色谱(LC-MS)结果证实该孵育产物为CRANAD-5。CRANAD-61不仅与H2O2作用迅速(图17-A),且灵敏度极高,能够检测出浓度低至5.0 nmol/L的H2O2(图17-B),同时对其他ROS(O2·-、·OH、OCl-、TBHP和NO·)也有着高灵敏度(图17-C)。此外,CRANAD-61的lgP=2.24,能有效穿透BBB。体内实验结果(图17-D)显示CRANAD-61静脉注射30、60、120及240 min后,AD脑部信号强度显著低于对照组,证明其监测AD脑中ROS变化的能力。
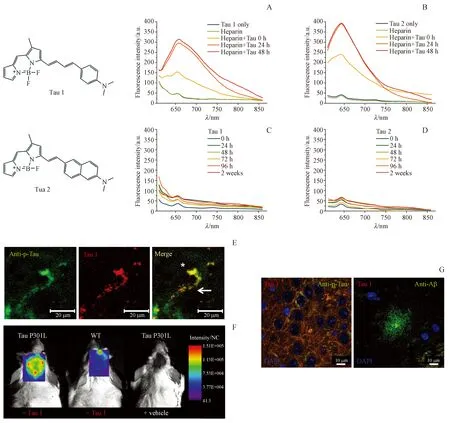
图15 (A,B)Tau 1和Tau 2与Tau蛋白和肝素孵育的荧光谱图;(C,D)Tau 1和Tau 2与Aβ纤维孵育的荧光谱图;(E)Tau 1的脑切片染色实验;(F) Tau 1的体内实验;(G)共聚焦成像实验[48]
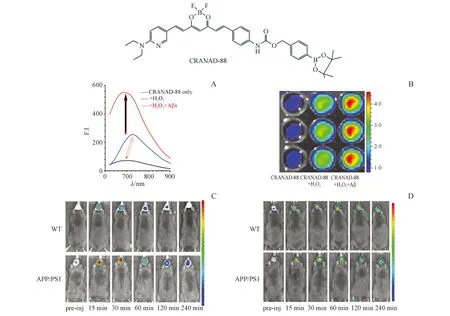
图16 (A)CRANAD-88与H2O2作用(一级放大),与H2O2+Aβ作用(二级放大);(B)级联放大验证实验;(C)CRANAD-88的体内实验;(D)加入丙酮酸钠(清除H2O2)的体内实验[54]
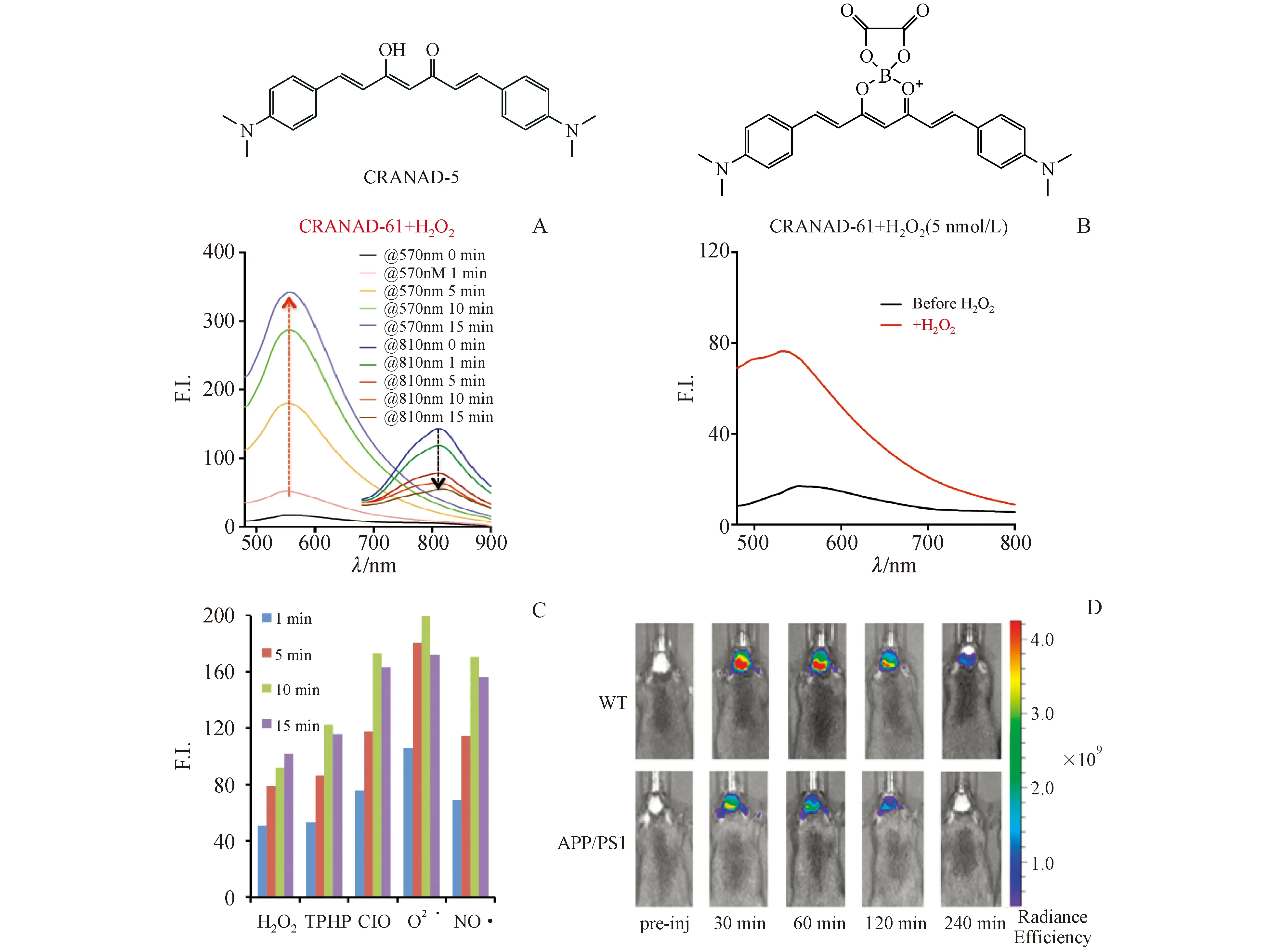
图17 (A)CRANAD-61与H2O2作用;(B)CRANAD-61与5.0 nmol/L H2O2作用;(C)CRANAD-61与不同种类ROS作用;(D)CRANAD-61的体内实验[55]
5 小结和展望
近年来,用于AD诊断的近红外荧光显像技术得到了迅速的发展,尽管已有多个探针分子的各项性质基本符合脑部成像的要求,但目前仍有许多问题和难点亟待攻克。
在上述所有探针中,Aβ探针占主要地位。研究表明,APP的水解和Aβ的生成始于AD早期,且先于Tau蛋白的聚合。但是,目前报道的探针绝大多数仅能用于标记Aβ斑块,而无法标记神经毒性更强的Aβ寡聚体。其次,Aβ斑块形成时对神经细胞已造成了不可逆的损伤,极大影响了AD治疗的效果。AD早期诊断技术的匮乏是AD治疗药物失败的一个重要原因。因此,开发能够特异性检测脑内Aβ寡聚体的NIRF探针是目前的当务之急,也是解决AD早期诊断和提高临床治疗成功率的有力保障。Ran课题组报道的探针CRANAD-102对sAβ的亲和力高于聚合物,但仍然无法区分单体和寡聚体。但其结构和设计思路对开发寡聚体选择性探针分子具有重要的指导意义。
针对Tau蛋白成像技术的研究不如Aβ成像研究详尽,仅有数个小分子探针被报道用于Tau聚合物成像。与一般的大脑成像荧光探针分子相比,Tau蛋白探针还需要具备高选择性(与Aβ结合能力低或不结合),给探针分子设计带来一定挑战。此外,Tau蛋白缠结形成于AD病理进程后期,Tau探针能否及如何用于AD早期检测还有待研究。
CRANAD-88和CRANAD-61为设计用于检测AD脑内ROS的NIRF探针提供了经验和范例。但是,氧化应激是众多疾病中极为常见的病理现象,ROS的过度积累在帕金森病、亨廷顿病等多种其他神经退行性疾病中也可检测到。因此,如何应用ROS探针进行特异性的AD诊断,区分AD和其他神经退行性疾病和脑部疾病是该领域的重点和难点。
