Gastrointestinal cancer stem cells as targets for innovative immunotherapy
2020-05-11MihaelaChivuEconomescuLauraNeculaLiliaMateiDenisaLauraDraguAnaNeaguIrinaAlexiuCoraliaBleotuCarmenCristinaDiaconu
Mihaela Chivu-Economescu, Laura G Necula, Lilia Matei, Denisa Laura Dragu, Ana I Neagu, Irina Alexiu,Coralia Bleotu, Carmen Cristina Diaconu
Abstract The role of cancer stem cells in gastrointestinal cancer-associated death has been widely recognized. Gastrointestinal cancer stem cells (GCSCs) are considered to be responsible for tumor initiation, growth, resistance to cytotoxic therapies,recurrence and metastasis due to their unique properties. These properties make the current therapeutic trials against GCSCs ineffective. Moreover, recent studies have shown that targeting stem cell surface markers or stemness associated pathways might have an additional off-target effect on the immune system.Recent advances in oncology and precision medicine have opened alternative therapeutic strategies in the form of cancer immunotherapy. This approach differs from classical anti-cancer therapy through its mechanism of action involving the activation and use of a functional immune system against tumor cells, instead of aiming physically destruction of cancer cells through radio- or chemotherapy. New immunological approaches for GCSCs targeting involve the use of different immune cells and various immune mechanisms like targeting specific surface antigens, using innate immune cells like the natural killer and T cells, T-cell chimeric antigen receptor technology, dendritic cell vaccine, or immune checkpoint inhibitors. In this respect, better understandings of immune regulatory mechanisms that govern anti-tumor response bring new hope in obtaining long-term remission for cancer therapy.
Key words: Immunotherapy; Gastrointestinal cancer; Cancer stem cells; CAR-T; Dendritic cells vaccines; Immune checkpoints inhibitors
INTRODUCTION
Gastrointestinal cancers include several malignancies of the gastrointestinal tract and accessory organs such as stomach, liver and intrahepatic bile duct, gallbladder and pancreas. All of them have epithelial cell origin, and combined accounts for 4974672 estimated new cases, representing 28% of all cancer incidence in 2018. According to GLOBOCAN 2018, all together gastrointestinal cancer are responsible for over 3.5 million deaths which correspond to 37% deaths of total human cancers[1]. Thus the need to understand the molecular background of gastrointestinal cancer, as well as mechanisms involved in occurrences and tumor maintenance, are of tremendous importance.
In the last years, the hypothesis that cancer appears from a population of stem cells has gained widespread support. Cancer stem cells (CSCs) are a distinct subpopulation within the tumor with unique properties. There are two theories about how tumors appear. Stochastic theory, involving the occurrence of unpredictable genetic and epigenetic changes during tumor growth[2,3]and hierarchical theory, which supports the idea of a subpopulation of cells, that have an intrinsic ability to initiate selfregeneration and tumor growth[4-6]. CSCs have been discovered in a wide field of tumors, including gastrointestinal cancer. These cells are at the origin of tumorigenesis and also, are responsible for tumor maintenance due to resistance towards standard oncology treatments, relapse, and metastasis[7]. More and more evidence is constantly accumulating that mechanisms of resistance of gastrointestinal cancer stem cells (GCSC) to conventional therapy are epithelial mesenchymal transition (EMT), drug efflux proteins, and upregulation of aldehyde dehydrogenase(ALDH) activity. Therefore, CSC raised an important challenge regarding the efficacy of current cancer treatment due to these special properties. In this way, more effective therapies that target the GCSCs subpopulation are needed, instead of addressing the entire tumor population. New immunological approaches involve the use of various immune mechanisms like targeting specific surface antigens or immune checkpoints on GCSC surface or using innate immune cells like the natural killer (NK) and T cells,T cell chimeric antigen receptor technology and dendritic cell vaccine (Figure 1).
TARGETING GCSC MARKERS
To target GCSCs markers and track the effect of anti-tumor therapies, it is necessary to identify them. Commonly, the surface antigens such as CD24, CD44, EpCAM, CD133,alone or in combinations, are among the most used markers for the identification of GCSC[8]. Combination of CD44, CD90, CD133 and ALDH1 was commonly used for esophageal tumor type[9,10], CD44 and ALDH1 for gastric tumor type[8,11], CD24, CD44,CD133, EpCAM and ALDH for pancreatic tumor type[12-14], CD44, CD90, CD133 and EpCAM for liver tumor type[15-18], CD24, CD44, CD49f, CD133, EpCAM and ALDH1 for colorectal tumor type[15,19,20](Table 1). Several other molecules, such as CD90,Musashi-1, LINGO2, oval cell marker OV6[8,21]have been reported as potential surface markers for GCSCs. Identification of specific antigens on the CSC surface may provide more targets for immunotherapy (Table 1).
These markers are relatively conserved across to the broad spectrum of solid cancers and also are common with normal stem cells[22]. Monoclonal antibodies,chimeric, humanized or fully human antibodies that are able to target specific markers were developed for the treatment of major malignant diseases, including gastrointestinal cancers (Table 2).
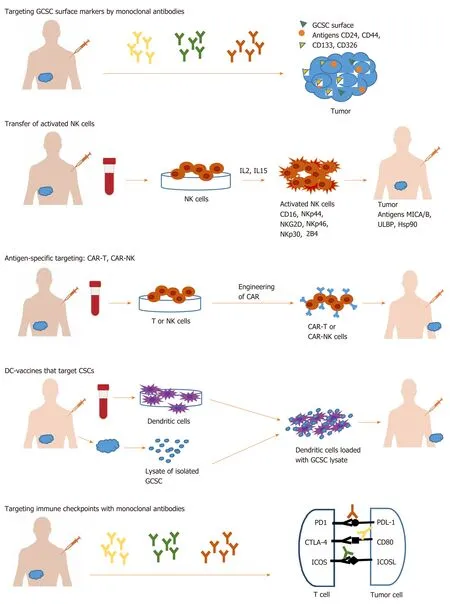
Figure 1 Immunological approaches for gastrointestinal cancer stem cell targeting. GCSC: Gastrointestinal cancer stem cell; NK: Natural killer; CSC: Cancer stem cell; CAR: Chimeric antigen receptor; DC: Dendritic cell.
CD44 is a transmembrane receptor commonly expressed on solid tumor CSC surface. Targeting CD44 with RG7356, a recombinant anti-CD44 monoclonal specific antibody, blocks the binding of CD44 to hyaluronic acid (HA) and inhibits cell adhesion, leading to tumor growth inhibition, and activates macrophages in preclinical models[23-25]. However, only a modest clinical efficacy was observed in patients with metastatic or locally advanced CD44-expressing solid malignancies(including breast cancer, melanoma, renal and lung cancer) treated with RG7356(phase I clinical trial)[26].
CD24 is a highly glycosylated protein localized in the membrane of many type of CSCs. Some CD24 monoclonal antibodies (mAb), such as SWA11and G7 mAbs, were developed for therapeutic purpose, demonstrating a good efficiency in human cancer xenograft models. A potent anti-tumor activity of SWA11 mAb[27]has been demonstrated in human colorectal cancer models, reducing tumor cell proliferation and angiogenesis[28]. Other anti-CD24 monoclonal antibody, G7 mAb was used in liver cancer xenograft models[29,30]demonstrating specificity for tumor tissue and efficacy in suppressing tumor growth, as single-agent treatment or in combination with doxorubicin[31], or cetuximab, a chimerical monoclonal anti-EGFR antibody[31].
CD326 (EpCAM) is a transmembrane glycoprotein mediating intercellular celladhesion in epithelial tissues being involved in cell signaling, proliferation,differentiation, invasion, metastasis, and chemo-/radioresistance. EpCAM is a target for immunotherapeutic strategies in epithelial-derived neoplasms of colon, stomach,pancreasetc.[32,33]. The antibody Catumaxomab (Removab®) targeting EpCAM was used in clinical trials on patients with ovarian, gastric, colon, breast cancers and malignant ascites (NCT00836654) delaying deterioration in quality of life for a prolonged survival period[34]. Moreover, one phase III clinical trial (NCT00822809)demonstrated that intraperitoneal infusion of catumaxomab activates immune cells such as NK cells, macrophages and T cells in ascites, and favors CD8+T cell accumulation into the peritoneal cavity showing a clinical benefits in treatment of malignant ascites associated with EpCAM positive carcinomas[33,35].
However, precise targeting with monoclonal antibodies seems difficult, since most GCSCs are identified based on a combination of surface markers, often unspecific[36].Since none of these markers is unique for GCSCs, additionally features of stem cells are such as ALDH activity, side population phenotype, the expression of pluripotency genes (OCT4, SOX2, and NANOG), and aberrant expression of ABC transporters[37]are analyzed in order to permit a more reliable identification and targeting of the cancer cells with stem properties[38,39].
Another widely used strategy against GCSC involved targeting of key signaling pathways like Notch, Hedgehog, Wnt, and IL6. However, accumulating evidence pointed out that the inhibitors used to target self-renewal pathways might have offtarget effects on immune cells, impairing T cell proliferation, function, and cytokine production[22,40-43].
At this point, scientists consider that cancer immunotherapy represents a new approach, different from the conventional one that uses chemo and radiotherapy.

Table 1 Gastrointestinal cancer stem cells markers
IMMUNOLOGICAL APPROACHES FOR GCSC TARGETING
Cancer immunotherapy differs from classical anti-cancer therapy through its mechanism of action involving the activation and use of a functional immune system against tumor cells, instead of aiming physically destruction of cancer cells through radio- or chemotherapy[44]. The immune system is an active part of the tumor microenvironment (TME). Here GCSCs co-exist with other cellular components like stromal cells, endothelial cells, immune cells such as dendritic cells (DC), NK cells, T cells, tumor-associated macrophages, regulatory T cells, tumor-infiltrating lymphocytes, and myeloid derived suppressor cells. There are several mechanisms used by GCSC in their interaction with TME to escape from tumor killer cells like NK and T cells. One of these refers to having a low expression of MHC class I surface molecules. Another one is represented by the crosstalk between GCSC and the other components of TME, these interactions being mediated by cytokines and chemokines that eventually suppress antitumor immunity. Moreover, recently, co-inhibitory molecules and immune checkpoint ligands such as programmed death-ligand (PD-L)1 and PD-L2 were identified to be overexpressed on GCSC surface. Due to PD1/PDL1 (L2) axis, GCSC can easily escape from immune cell action. Understanding the TME and the dynamic cross talk between GCSCs and the TME is equally important to initiate an efficient anti-tumor therapy without impairing the anti-tumor immune response.
Immunological approaches for GCSC targeting involve different immune cells and various immune mechanisms like using innate immune cells such as NK and T cells,using antigen-specific targeting by T cell chimeric antigen receptor (CAR) technology,DC vaccine, or immune checkpoints therapy[45,46]. The targeting strategies against GCSC are listed in Table 3.

Table 2 Targeting gastrointestinal cancer stem cell surface markers by monoclonal antibodies
NK transfer in cancer immunotherapy
NK cells, the third largest population of immune cells after B and T lymphocytes,serve the innate immunity, usually defending the human organism against infections.NK are good candidates for immunotherapy since they trigger special attacks on cancer cells that express ligands that couples activating receptors on NK cells. This action is mediated through a group of activating receptors containing CD16, NKG2D,NKp30, NKp44, NKp46, 2B4 and DNAM-1 with PVR and NECTIN-2[47-50]. The major activating ligands for NK cells are MICA/B, ULBP and Hsp90 usualy overexpressed on tumor cells[51]. For tumor eradication is necessary total destruction of CSCs.Different studies showed that there are CSCs that express ligands that can be recognized by NK cells and, consequently can be killed[52-54], and certain CSCs which do not show detectable ligands for NK and escape cytotoxicity[55]. Anin vitrostudy conducted by Ronget al[56]showed that cytokine-induced killer cells, which are NK lymphocytes characterized by the co-expression of CD3 and CD56 surface antigens,killed CSCs in hepatocellular carcinoma via interaction of their membrane receptor NKG2D with stress-inducible molecules, MIC A/B and ULBPs, on target cells.In vivo,cytokine-induced killer infusion significantly delayed tumor growth. Similarly, Ameset al[57]demonstrated that activated NK cells are capable of preferentially killing tumor cells with a CSC phenotype identified by multiple markers (CD24+/CD44+, CD133+,and aldehyde dehydrogenasebright) from a wide variety of human cancer cell lines,including pancreatic cell lines like PANC-1 and BXPC3. The mechanism of action implicated an NKG2D-dependent NK activation via MICA/B, Fas, and DR5 ligands expressed on GCSCs. Also, Yinet al[58]showed that cells with stem cell phenotype can be more easily killed by NK cells activated by IL2 and IL15. Taken together, these preliminary studies provide evidence that activated NK cells can have translational potential as NK immunotherapy against GCSC phenotype, and other CSC from solid malignancies.
Antigen-specific targeting by T cells, CAR-T, CAR-NK
The next step in cancer therapy resided in the generation of effector T cells and NK cells genetic engineered to produce an artificial T cell receptor, named CAR that gives T cells both the ability to target a specific protein/tumor antigen and to be consequently activated. CAR-T cells against GCSC antigens have been developed and evaluated in several gastrointestinal cancer models (Table 3).
Miyamotoet al[59]used CAR-T-based immunotherapies against colorectal CSCs based on the ASB4 gene that was identified as being expressed on colorectal CSC, but not on non-CSC cells or normal cells/tissue. ASB4 as tumor-associated antigen was used to generate specific cytotoxic T lymphocytes (CTL)in vitro, that were able to infiltrate implanted colorectal tumors in a mouse model, preventing tumor growth.Another clinical trial was developed by Fenget al[60]that used CAR-T cells targeting epidermal growth factor receptor (EGFR) and GCSC surface antigen CD133,respectively. The patient received successive infusions of CAR-T cells for the treatment of unresectable/metastatic cholangiocarcinoma. The results showed a partial response of 8.5 mo from CAR-T for EGFR and 4.5-mo-lasting from the CAR-T for CD133 treatment, however, both therapies induced acute toxicities. Maliaret al[61]developed CAR-T for CSC antigen CD24 and evaluated it in mice with pancreatic adenocarcinoma xenografts. The results showed that more than 50% of the animals remained tumor-free after 16 wk.
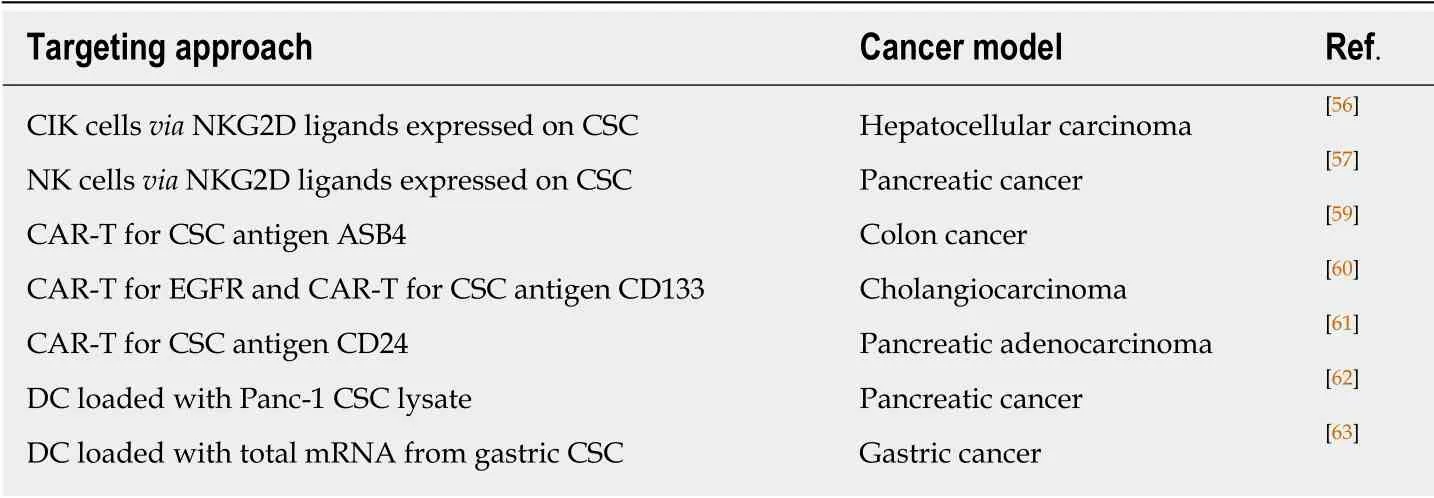
Table 3 Targeting gastrointestinal cancer stem cell by natural killer cells, chimeric antigen receptor expressed on T cells and dendritic cells based vaccines
As concluding remarks, treatment of solid tumors using CAR-T cells induced less favorable results then hematological malignancies, mainly due to short efficacy and off-target toxicity. Davenportet al[62]reported in 2018 that responses triggered by CAR-T cytotoxic cells are fast but short, being followed by a rapid loss after 20 h in cytotoxic activity against tumor. The main concern remains on the severe toxicity associated with CAR-T therapy like cytokine release syndrome, which can trigger organ damage and death, neurologic toxicity, insertional oncogenesis, graft versushost disease, off-target antigen recognition[63]. Two trails using CAR-T cells engineered against ERBB2 or CEACAM5 used for the treatment of gastrointestinal cancers reported poor efficacy and caused acute pulmonary toxicity due to antigen expression on lung epithelium. This resulted in the death of one patient within 5d post-transfer of the cellular product due to multiple organ failure[64,65].
In order to avoid CAR-T therapy accompanied toxicity is essential to choose accurate target antigens and improve tumor discrimination[66,67]. Also, there are some studies on introducing a suicidal gene that can induce apoptosis of T cells to prevent over activation and critical off-tumor cytotoxicity. Such genes are the thymidine kinase gene of herpes simplex virus and inducible caspase 9. If the latter seems successful, the first strategy, related to the thymidine kinase gene of herpes simplex virus seems to raise immunogenicity problems and it will not be used in the clinic[67-69].
Recently, the knowledge of producing CAR-T has been transferred to CAR-NK cells. CAR-NK use seems to be safer in the clinic, as NK cells do not initiate similar toxicity[70]. Between the advantages: CAR-NK cells are able to retain the expression of their activating receptors, showing longer efficacy, and appear safer in terms of cytokine release syndrome and neurotoxicity due to a different pattern of cytokines/chemokines released after activation[71,72]. However, there are also major drawbacks like the poor ability of NK to reach tumor tissue due to TME and some changes that may intervene in the expression of activating receptors/ligands. For example, the level of NKG2D ligand is increased in the early stages of colorectal cancer, but it decreases during tumor progression[73].
There are not ongoing clinical trials for GCSC or other solid CSC. The only clinical trial for solid cancers is a phase I study that use anti-GD2 CAR-T for sarcoma and neuroblastoma patients (NCT02107963), without published results. There are however 30 clinical trials that are recruiting patients with solid cancers including liver, stomach and colorectal for CAR-T therapies using PD-1, CTLA4, EGFR or NKG2D-ligand.Also, there are three trials for solid cancers that are recruiting for CAR-NK therapies using antigens like MUC3, ROBO1, and NKG2D-ligand. The creation of NKG2D/NKG2DL-based multi-functional fusion proteins is becoming one of the most promising strategies in immunotherapy for cancer. Activated CAR-NKG2D receptor on the T or NK cell surface can bind to its respective NKG2D ligand expressed in tumor cells, enabling immune cells to kill tumor cells.
DC-vaccines that target GCSC
Dendritic cells are crucial players in immune responses, and their ability to control the activation, proliferation and differentiation of specific T cell subsets make them strategic tools for cancer vaccines that target CSCs. A great advantage of DC-vaccines might be the potential capacity to induce immunological memory, eliminating existing CSCs and, at the same time, offering long-term protection against new arising CSCs[74].
Most clinical trials using DC-vaccines are based on DC loaded with lysates of isolated CSCs. Yinet al[75]used such DC loaded with pancreatic Panc-1 CSC lysate and observed that modified DC induced proliferation of T cell lymphocytes during coculture. This approach has at least a few disadvantages like the lack of reliable surface makers that may be used for CSCs isolation, unknowing which neoantigens in the lysate elicit an immune response, and the variability of the number of immunogenic neoantigens on different types of tumors[76,77].
ALDH, a marker frequently used for CSC identification was used in obtaining DC vaccines that significantly inhibited tumor growth, reduced development of pulmonary metastases and prolonged survival. Direct targeting of CSCs was confirmed by the specific binding of IgG produced by ALDHhighCSC-DC vaccine primed B cells to ALDHhighCSCs, lysing these target CSCs in the presence of complement[78]. This promising approach was reported so far for squamous cell cancer and metastatic melanoma, however, ALDH is a highly expressed marker on various GCSC (Table 1), so we can hypothesize similar positive results in gastrointestinal cancers.
In spite of the promising results, DC-based vaccination strategies need to be improved[78]. A more efficient strategy to eradicate CSCs might be to load autologous DCs with peptides, proteins or even mRNA rather than tumor lysate, this way controlling more accurately the generated immune response. In this regard, Bagheri Vet al[79]loaded DC with total mRNA from gastric CSC expressing CD44, CD54, and EpCAM markers. These DC were able to induce IFN-γ gene expression in Tlymphocytes after a 12-d co-culture.
Recent studies proposed loading DCs with transcription factors as NANOG,OKT4a, SOX2, c-MYC, and KLF4, which also transform somatic cells into stem cells(iPS). Targeting CSCs unique proteins might be one of the best ways to destroy CSCs.Since the expression of NANOG is low or absent in normal cells and CSCs re-express it, makes NANOG an ideal therapeutic target. DCs loaded with NANOG peptides will be able to generate immunological memory after vaccination and would help the immune system to manage CSC plasticity[22].
In order to maximize the response rates to vaccines, if NANOG peptides cannot be presented by a patient HLA class I molecules, peptides against other stem cell transcription factor OKT4 or SOX2 may be used[76]. Combining DC vaccination against CSCs with other therapies, as immune checkpoint inhibitors, for example, might overcome immunosuppressive mechanisms in cancer and avoid bone marrow damaging by the chemotherapeutics.
IMMUNE CHECKPOINTS
It is considered that CSCs escape immune surveillance due to their immune suppressive profiles based on the expression of co-inhibitory molecules, immune checkpoints ligands and cytokines, drug-resistance and ability to activate an EMT programme[80]. Based on these properties, CSC not only escapes immune surveillance but also directly inhibits T and NK cells anti-tumor activityviamodulating immune checkpoints.
Several immune checkpoints have been stated during last years with either costimulatory activity on immune cells such as CD28/CD80 (CD86), ICOS(CD278)/ICOSL, CD27/CD70, GITR/GITRL, or co-inhibitory like PD-1/PDL-1 (PDL2), BTLA/HVEM, CTLA4/CD80 (CD86), B7H3, B7H4, B7H5/HVEM, LAG3/MHC II, TIM3/GAL9, TIGIT/Nectin-2, or IDO. Many of them are highly expressed on various CSCs, but the type of molecule seems to vary with tumor type and localization.
From these, PD-L1 (also known as CD274 or B7H1) and B7H3 have been identified as promoters of CSC-like phenotype, EMT, tumor cell proliferation, metastasis and resistance to therapy[81-83].
PD-L1 is one of the most studied immune checkpoints. The interaction between PDL1/PD-L2 and PD-1 aids CSCs in escaping from the killing through inhibiting tumorreactive T cells by binding to its PD-1 receptor. Moreover, PD-L1 is also expressed by tumor-associated myeloid-derived suppressor cells, contributing to T cells blocking and immune deficiency in TME[84]. Hsuet al[85]established that PD-L1 high expression in CSCs is due to EMT and to EMT/β-catenin/STT3/PD-L1 signaling axis. Moreover,PD-L1 expression could be enhanced via PI3K/AKT and RAS/MAPK pathways. All these major pathways could be activated by OCT4 and SOX2, key regulatory genes involved in CSC self-renewal and function[86]. The final effect of PD-L1 overexpression on CSC will be an increase in cancer invasion and proliferation via EMT.
This hypothesis was sustained by several experiments on GCSC. Yanget al[87]detected PD-L1 overexpression on gastric CSCs, defined as Lgr5+/CD326+/CD45-,were enhancedin vitrotumor-promoting capacity of GCSCs by colony-forming assay,and induces their proliferation. In reverse, knockdown of PD-L1 expression in gastric cancer cells significantly suppressed proliferation and invasionin vitro[88], and tumor growth in nude mice[89].
An increased level of PD-L1 was observed in esophageal and colorectal CD133+GCSCs with EMT phenotype. The authors showed by manipulating PD-L1 expression, that higher PD-L1 expression promoted cell proliferation, migration and EMT phenotype. The EMT mechanism could help GCSC escape immune attack during metastasis[90].
The assessment of PD-L1 level on biopsies could bring useful information for establishing therapies regimen. The dynamic change of PD-L1 expression may indicate the response to therapy and have predictive significance on progression free survival. This could be monitored with the help of circulating tumor cells, which may act as substitute for tissue biopsies, and have great utility in real-time cancer management[91].
The expression of these molecules with an immunosuppressive effect on the GCSC surface may be a major problem as cytotoxic T lymphocytes therapies become less effective. However, is an indicator that GCSC resistant to classical anti-tumor therapy could be targets for immune checkpoints inhibitors.
Targeting immune checkpoints with monoclonal antibodies has become a custom treatment since early studies have shown their ability to improve tumor infiltration of CD8+and CD4+T cells and inhibit tumorigenesis.
Antibodies blocking PD-L1, PD-1 or CTLA-4 were developed and tested in clinical trials for their cancer therapeutic potential. 2014 was the year of nivolumab and pembrolizumab approval, both being monoclonal antibodies targeting PD-1. They were authorized for clinical use with benefits in various types of cancer such as refractory malignant melanoma[92], Hodgkin lymphoma[93], NSCLC[94], head and neck squamous cell carcinoma[95], urothelial cancer[96], gastric adenocarcinoma[97], colorectal cancer[98]and advanced hepatocellular carcinoma[99](Table 4). Moreover, pembrolizumab has received general approval for the treatment of all solid tumors with high microsatellite instability and deficiency in the mismatch repair genes. Next, a combination of ipilimumab (anti-CTLA-4) with nivolumab (anti-PD-1) inhibitors was shown to significantly enhance antitumor efficacy and the response rates in patients with colorectal cancer expressing high microsatellite instability phenotype.
In addition to targeting PD-1 and CTLA-4 receptors, PD-L1 has been also confirmed to be useful for immunotherapy. It was demonstrated that increased expression of PD-L1, decreased T cell infiltration and activation, protecting tumor and GCSCs against immune response. In 2016 atezolizumab, an anti-PD-L1 monoclonal antibody received approval for the treatment of several solid cancers, but not yet for gastrointestinal cancers.
The clinical benefits of immunotherapy cannot be questioned. The development of new inhibitors for immune checkpoints or their ligands continues, addressing newly identified regulators Lag-3, Tim-3, TIGIT, V-domain Ig suppressor of T cell activation,etc. And this is only the beginning, as many clinical trials are under way to assess the effectiveness of combining these inhibitors either with or without classical chemotherapy in treating gastrointestinal cancers[100].
COMBINATION IMMUNOTHERAPY
To target GCSC and completely eradicate them, it might be necessary to combine these immunotherapy approaches.
Several clinical trials are now proposing interesting strategies for combining immune checkpoints therapy with DC-vaccines or CAR-T technology. Most of these clinical trials are either in the phase of patient recruitment or in phase I. There are several trials that are testing the combination between anti PD-1 compound(nivolumab) and gene-modified T-cells and dendritic cell vaccine targeting cancertestis antigen (CTA). CTAs such as New York esophageal squamous cell carcinoma 1 and melanoma-associated antigen A (MAGEA) are considered excellent candidates for cancer immunotherapy since a large majority of them have their espression limited to the embrionic stem cells, testes, ovaries and endometrium in normal tissue, and are re-expressed in metastatic tumours[101]. MAGEA1-3, MAGEA9, LAGE1, and New York esophageal squamous cell carcinoma 1 were found to be highly expressed in hepatocellular carcinoma, oesophageal, gastric and colorectal carcinoma stem/progenitor cells and associated with poor survival, high risk of tumor recurrence[102,103].However, there is a small number of CTAs that are expressed on normal tissue as well. Although targeting CTA seems to be a promising strategy, carefull selection of CTA type for immunotherapy is manadatory. Serious neuronal adverse events followed by death were observed during clinical trials using anti-MAGE3 CAR-T cells in patients with solid cancers. Histopathological examination showed that normal neuronal cells also expressed MAGEA proteins that became targets of modified T cell therapy, thus being destroyed[104].
Another approach is targeting of inhibitory immune checkpoints like PD-1 and TIGIT to unblock the NK and T cells activity. Zhanget al[105]found that blockade of TIGIT promoted NK cell mediated immunity in a mouse model with colon cancer,and the response was enhanced by additional anti-PD-L1 immunotherapy. Moreover,it seems that the animal model developed a persistent memory immunity that was functional after tumor cell reinfusion.
Combined immune therapies may be very effective having the advantage of addressing both GCSC and TME simultaneously. Thus, they can target, for example,GCSC surface markers with monoclonal antibodies, DC-vaccines or CAR-T therapy,and at the same time, they can reactivate the immune system by blocking the negative signals induced by immune checkpoints in effector immune cells. However, there are some limitations since most of the known solid tumor-associated antigens are expressed also in normal tissues, resulting in damaging off-target toxicity. Therefore,there is a continue effort to identify tumor-specific antigens that can be addressed using immune therapies. Another limiting factor that can influence the clinical response is the level of inflammatory infiltration and the expression of immune checkpoints. Unlike liquid cancers, where immunotherapy has been a real success, in solid tumors, their efficiency has been diminished by the consistency, content and dynamics of TME that modulates the anti-tumor response through access and phenotype of immune cells.
To improve the efficiency and ensure the safety of the treatment it is imperative to carefully select the target antigens, assuring that they are highly immunogenic and expressed only in targeted the cell population.
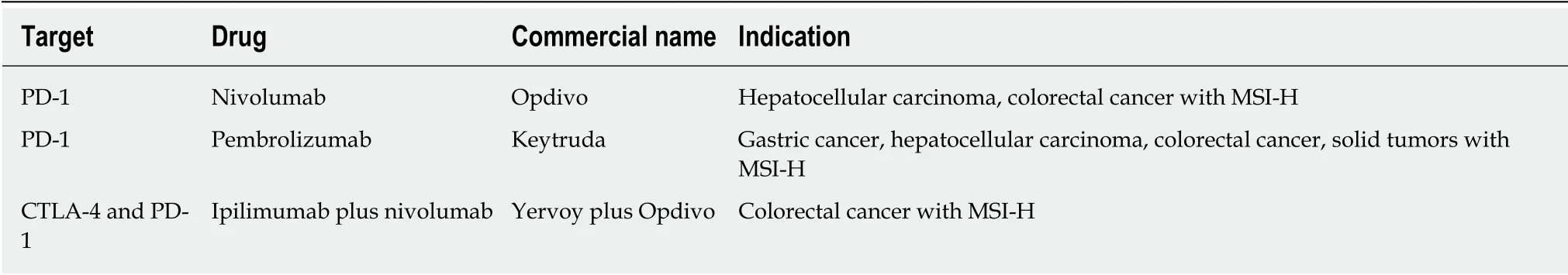
Table 4 List of approved drugs targeting immune checkpoints for gastrointestinal cancers
CONCLUSION
Tumor-immune profiling has highlighted the mechanisms of immune evasion of cancer based not only on CSC properties but also on the interaction of these cells with TME. These include features such as antigen presentation and regulation of immune cells activation and functioning through immunosuppressive elements like immune checkpoints. Novel immunotherapeutic approaches addressed to all these features.There are several approaches that involve expanding of NK and T cells for CSC antigen-specific targeting or dendritic cell-based vaccines against CSCs. However, the most exciting approach is related to immune checkpoints discovery. Targeting PD-1,CTLA-4, Lag-3, Tim-3, and TIGIT, or their respective ligands on CSC allows activation of the immune cells like T-lymphocytes, NK, neutrophils, dendritic cells and destruction of CSC. The main purpose of these approaches is to modify the TME so that tumor cells and CSC become more responsive to chemotherapy.
An important limitation may come from gaining resistance to immunotherapy. It may be caused by the absence of tumor antigens, loss or decrease of MHC expression,alteration of signaling pathways affecting immune cell infiltration, or presence of regulatory T cells or myeloid derived suppressor cells in the tumor microenvironment. In order to prevent resistance and extend the clinical benefits of immunotherapy, it is necessary to better understand the anti-tumor response mechanisms of the strategies discussed here in order to combine them, as combinatorial therapy might be the answer for acquiring long-term remission in cancer therapy.
杂志排行

World Journal of Gastroenterology的其它文章
- SARS-COV-2 infection (coronavirus disease 2019) for the gastrointestinal consultant
- Optimized timing of using infliximab in perianal fistulizing Crohn's disease
- Intestinal epithelial barrier and neuromuscular compartment in health and disease
- Is the measurement of drain amylase content useful for predicting pancreas-related complications after gastrectomy with systematic lymphadenectomy?
- Silymarin, boswellic acid and curcumin enriched dietetic formulation reduces the growth of inherited intestinal polyps in an animal model
- Lifestyle factors and long-term survival of gastric cancer patients:A large bidirectional cohort study from China Zhao LL,