虫草素衍生物修饰纳米金的制备及其生物活性研究
2020-05-04崔琳琳雒江菡魏思亦
崔琳琳,王 莹,雒江菡,袁 甜,魏思亦
(1.哈尔滨商业大学药学院,黑龙江哈尔滨 150076;2.黑龙江省预防与治疗老年病药物研究重点实验室,黑龙江哈尔滨 150076)
虫草素(3′-脱氧腺苷)是1951年经过Wu[1]等研究,从人工栽培的蛹虫草的培养液中分离得到的一种核苷类似物。目前,虫草素因其广泛的药理作用而受到了众多学者的关注,现已成为抗衰老、保健、新药研发等领域的热门研究内容。现有研究结果证明了虫草物种具有抗氧化、抗炎、抗肿瘤、免疫调节和肾脏保护等药理作用[2-6]。纳米金作为优良的载体,由于其具有表面易修饰性,通过络合到其他化合物结构上增加其药理活性,被越来越广泛用于生物医学研究中[7-13]。通过小分子络合纳米金可以减少传统抗癌药物对正常细胞的毒性,增加其抗癌作用效果[14-16]。纳米金颗粒通过不同配体修饰后,使之可应用于检测、靶向治疗以及生物等一系列领域[16-18]。近年来,虽然国内外研究人员对虫草素进行了一些研究,但是其药理作用机制的研究还不够深入,将虫草素作为纳米金的小分子修饰物方面尚缺少尝试。虫草素结构中含有-OH,故本实验尝试利用其结构中-OH修饰至纳米金的表面,以期开发具有较高药理活性的新型药物。

图1 化合物的合成路线Fig.1 Synthetic routes of compound
在本文的研究中,以虫草素为母核,在5′位置上接入新的基团,制备了一个虫草素衍生物(化合物B),通过提高脂溶性增加其活性。将化合物B作为小分子,制备了其修饰的纳米金络合物C。通过MTT实验研究对HepG2细胞的抑制作用,并以革兰氏阴性菌大肠杆菌,革兰氏阳性菌枯草杆菌和金黄色葡萄球菌为研究对象,通过抑菌圈法进行抑菌活性研究,采用浊度法确定其最小抑菌浓度。以期为虫草素衍生物抗肿瘤以及抑菌作用的深入研究和探索虫草素衍生物在肿瘤治疗应用方面的前景提供依据。虫草素衍生物修饰纳米金为今后研究小分子修饰纳米金的合成手段提供新的思路。
1 材料与方法
1.1 材料与仪器
虫草素原料药 纯度>99%,湖北邦盛化工有限公司;2-呋喃甲酰氯 上海阿拉丁生化科技股份有限公司;吡啶 分析纯,天津富宇精细化工有限公司;二氯甲烷 分析纯,天津天力化学试剂有限公司;甲醇 分析纯,天津天力化学试剂有限公司;氘代试剂-DMSO 北京金鸥翔;蒸馏水 实验室自制;3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT) 北京欣经科生物技术有限公司;杜尔贝科改良的伊格尔培养基(DMEM) Hy Clone/Thermofisher;胎牛血清(FBS) 杭州四季青生物有限公司;Hank’s平衡盐溶液和胰蛋白酶消化液 GIBCO公司;人肝癌HepG2细胞株 中国医学科学院基础医学研究所细胞中心;氯金酸 天津迈斯科;碳酸钠 天津天力化学试剂有限公司;溴化钾 天津天力化学试剂有限公司。
布鲁克核磁共振波谱仪(500 MHz) 布鲁克(北京)科技有限公司;UV-5200 PC紫外可见分光光度计 上海元析仪器有限公司;TECHAI-10透射电子显微镜 荷兰PHILIPS;AL-Y3000X射线衍射仪 丹东奥龙射线仪器基团有限公司;FTIR-650傅里叶变换红外光谱仪 天津港东科技发展股份有限公司;高压灭菌锅 日本Sanyo公司;DL-CJ-1ND 医用型超净工作台 中国北京东连哈尔仪器制造有限公司。
1.2 实验方法
1.2.1 化合物的合成 称取2.259 g(0.009 mol)虫草素(化合物A)溶于200 mL脱水吡啶中(40 kHz超声波助溶5 min)。在磁力搅拌和氮气保护下,分别将0.01 mol 呋喃缓慢滴入虫草素的吡啶溶液中。保持温度60~70 ℃,TLC监测反应进程,并在原料消失后终止。 然后通过旋转蒸发器在95 ℃下将反应溶液浓缩成浓膏状[19]。分别用30 mL二氯甲烷和水液混合,萃取三次,合并有机相,将上述合并的有机相用旋转蒸发仪减压蒸馏得到固体粉末样品,采用干法上样的方法,用少量硅胶粉末拌匀样品,柱层析(甲醇∶二氯甲烷=1∶15),上硅胶柱(2 cm×75 cm玻璃柱,200~300目柱层析硅胶)得到化合物B。
在4 ℃预冷的40 mL的双蒸水中,加入0.6 mL质量分数为1%的氯金酸;再加入0.2 mol/L的碳酸钠溶液0.2 mL;在搅拌的条件下,迅速分批次加入新鲜配制的化合物B水溶液(质量浓度为0.5 mg/mL)0.4 mL,直至溶液出现酒红色为止,磁力搅拌5 min,4 ℃静置保存,得络合物C溶液。
化合物产率计算:产率(%)=实际产量/理论产量×100
1.2.2 化合物的鉴定和表征
1.2.2.11H NMR和13C NMR鉴定化合物B的结构 二甲基亚砜(DMSO)用作溶剂溶解化合物A和化合物B,均以四甲基硅烷(TMS)为测定内标,通过500 MHz1H NMR和13C NMR对化合物B的结构进行确认。观察化合B新增或减少的质子峰的种类和数量与目标化合物的吻合程度,从而判定新化合物B的结构。
1.2.2.2 分子量测定 将化合物B用色谱级乙腈超声溶解,制备成浓度为100 μg/mL的溶液,进行质谱测定,确定分子量。质谱分析条件:电喷雾离子源,正离子扫描模式,毛细管电压3.5 kV,喷雾电压5500 V,雾化气压力0.11 MPa,辅助气柱前压68 kPa,干燥温度450 ℃,干燥气体流速为8.0 L/min。
1.2.2.3 红外光谱分析 使用干燥的KBr压片,傅立叶红外分光光度仪测定虫草素衍生物和虫草素红外光谱,观察新化合物红外色谱峰的变化情况,对化合物A、B和C进行表征。
1.2.2.4 UV光谱分析 以甲醇为空白对照,超声助溶,对络合物C进行紫外扫描(波长范围0~600 nm),观察络合物C吸收波长的变化情况。
1.2.2.5 X射线衍射分析 将络合物C溶液100 ℃烘干成干燥颗粒状态后平铺在载玻片上,进行X射线衍射图(XRD)图谱测定。
1.2.2.6 透射电子显微镜分析 使用透射电子显微镜(TEM)检查胶体金颗粒的尺寸,将络合物C滴在涂有碳的铜网上并真空干燥,然后在TEM下观察。使用氯金酸还原的方法。 氯金酸被还原成金原子,由许多金原子组成积聚成胶体金溶液。利用TEM进行检测,TEM图像表明络合物C颗粒的分散程度。
1.2.3 化合物的体外活性研究
1.2.3.1 体外抗肿瘤活性研究 细胞培养[20]:HepG2细胞解冻后,放入超净工作台,加入离心管内并加含有10%胎牛血清的DMEM培养液,混匀后1500 r/min离心20 min。弃去上清液,向离心管加入培养液,吹打制成细胞悬液。细胞计数,调细胞浓度为3×104cell/mL。将培养瓶放入37 ℃,5% CO2培养箱内培养,进行常规培养传代。
MTT实验:在体外抗肿瘤活性研究中,以人肝癌细胞HepG2为研究对象。采用MTT法检测不同浓度的化合物A,化合物B和络合物C对HepG2细胞增殖的影响。根据文献方法[21],化合物A、B、C均制备成浓度为80、40、20、10、5、2.5、1.25 μg/mL的溶液。细胞以 3×105个/mL接种量接种于96孔培养板,5% CO2,37 ℃温箱中培养48 h后,每孔加入不同浓度的药物各100 μL,继续培养24 h、48 h,每孔6个平行孔。观察到培养液变黄且细胞未长满时,更换培养液。分别加入100 μL MTT(噻唑蓝)后放回培养箱继续培养4 h弃去培养基,加入150 μL DMSO,在酶标仪570 nm下读板,根据测得的吸光度值,计算细胞生长抑制率,用分析软件SPSS计算IC50值。
1.2.3.2 体外抑菌研究 抑菌实验:含药纸片的制备[22]:分别将大肠杆菌、枯草杆菌、金黄色葡萄球菌活化并将活化菌液浓度调制为1×108CFU/mL取100 μL加入灭菌后的固体培养基上,直径为6 mm的圆型纸片灭菌后分别浸泡在化合物A、化合物B和络合物C中。取出沥干后放置平板上。37 ℃恒温培养箱培养24 h。观察结果,测定抑菌圈直径。每组实验重复3次。
络合物C对三种菌的最小抑菌浓度实验[23]:将络合物C稀释到无菌水中,制备成600 μg/mL原液,过滤后使用。二倍法稀释药物浓度。菌液浓度为1×108CFU/mL。将100 μL的含药溶液加入到96孔板中每行的1~11孔内,第12孔不加药物做空白对照。为了避免交叉污染,每块96孔板只接取一种菌,为了降低边缘效应,其他孔均加入100 μL无菌水和100 μL营养肉汤培养液。每组实验须做三组重复。将96孔板放置恒温培养箱37 ℃摇床孵育24 h判断结果,酶标仪测定OD600。
1.3 数据处理
实验中增值率用百分数表示,通过 SPSS 17.0 软件进行统计学处理,相应的各组间比较采用LSD-t检验,P<0.05差异有统计学意义。
2 结果与分析
2.1 化合物B的结构
所得到的化合物B为白色固体粉末,熔点197~198 ℃,产率为50.4%。
1H NMR(500 MHz,DMSO-d6)δ:8.26(1H,s,2-H),8.13(1H,d,8-H),7.98-6.70(3H,d,5′-CH=),5.93(1H,d,1′-H),5.77(1H,d,1′-H),4.69-4.24(4H,ddtd,2′-OH,4′-H,5′-CH2),2.06(2H,ddd,3′-CH2);13C NMR(126 MHz,DMSO-d6)δ:158.16(1C,-COO),156.52(1C,6-C),153.10(1C,2-C),149.47(1C,4-C),148.27-143.97(2C,5′-CH=),139.31(1C,8-C),119.44(1C,5-C),119.15-112.85(2C,5′-CH=),91.22(1C,1′-C),77.87(1C,4′-C),74.81(1C,2′-C),65.95(1C,5′-C),35.17(1C,3′-C). [M+H]+=346.1154 m/z.
2.2 络合物C的结构
化合物A、B和C的红外光谱如图2所示,化合物A的红外吸收峰:3141 cm-1是羟基的特征吸收峰,3337 cm-1是N-H的伸缩振动吸收峰,1341 cm-1是5′伯醇的羟基吸收峰[24]。 在图中,化合物B的红外吸收峰,产物在1740 cm-1处显示出强吸收峰,而A在1740 cm-1处没有明显的吸收峰,这是5′羟基的特征吸收。在该图中,络合物C的红外吸收峰,3455 cm-1是N-H的伸缩振动,1632 cm-1是C=N伸缩振动。C的峰值由于纳米金偶联的影响,峰值发生偏移。
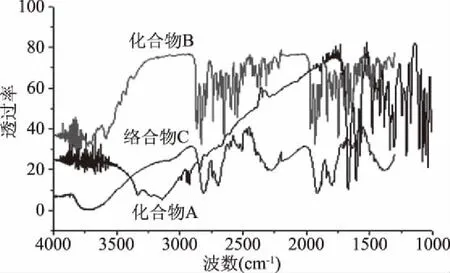
图2 红外光谱图Fig.2 IR spectrum
络合物C的UV光谱如图3所示。在200~600 nm的波长范围内扫描。这与纳米金溶液的吸收特性最大吸收峰长为521 nm一致[25],可以确定复合溶液含有纳米金颗粒。

图3 络合物C的UV光谱Fig.3 UV spectrum of complex C
络合物C的X射线衍射分析结果见图4。与JCPDS标准卡对照,可以看出样品特征峰的2倍衍射角2θ值依次为5.70°、32.30°、46.20°处,出现的3个衍射峰均为Au的特征衍射峰,分别与面心立方(FCC)结构AuC(111),(200)和(220)晶面相对应,由此确定纳米金。在不同放大倍数镜像下观察络合物C的形态,结果见图5所示,从图中可见,粒子外观圆整,大小均匀,分散性好。金颗粒为球型,基本不团聚。根据研究发现[26-27],Au簇自身具有电子结构,Au簇通过掺杂正电性元素或通过与稳定分子的亲核位点与化合物所带-OH官能团相互作用。由于氧阴离子在Au表面上的吸收增强了电子密度促进氧分子的吸收,自组装单层包覆的Au-NPs与羟基进行吸附,Au核心的大量电子电荷转移吸附到-OH上,从而Au和-OH基团之间形成复合物。由此推测化合物B中羟基为与纳米金可能的络合位点。
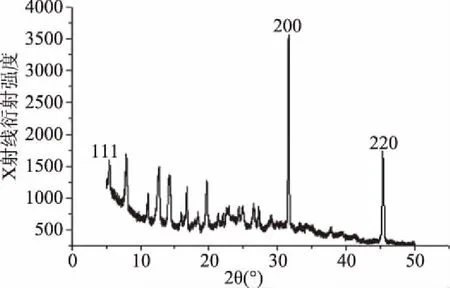
图4 络合物C的XRD Fig.4 XRD of complex C

图5 络合物C的TEM照片Fig.5 TEM photo of complex C
2.3 生物活性结果
图6为三个化合物对HepG2细胞抑制率随浓度变化曲线。如下图所示,在一定浓度范围内肿瘤抑制率的强度随着药物浓度的增加而增大。当药物浓度为40~80 μg/mL时,络合物C的抑制作用明显优于化合物B。化合物A化合物B以及络合物C的IC50值分别为39.41,39.01和36.61 μg/mL。在药物浓度高于40 μg/mL时,络合物C的抑制率明显高于化合物A和化合物B,说明与纳米金络合后的络合物C对HepG2细胞作用提升明显。三个化合物对HepG2细胞均有抑制作用,其中络合物C的抑制效果最佳。
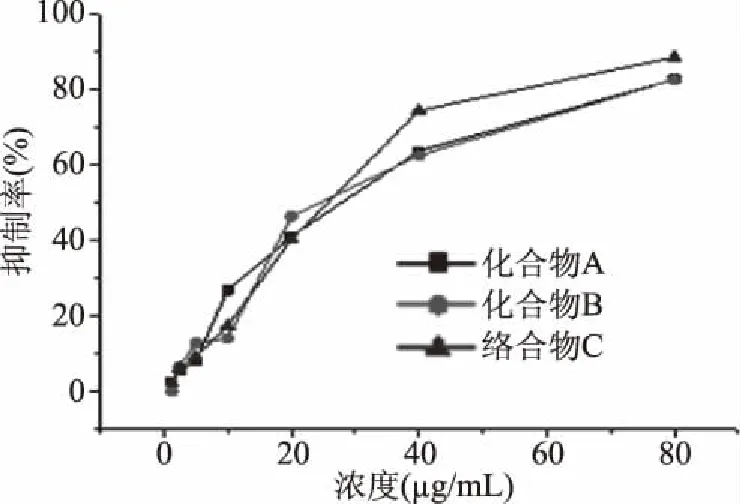
图6 药物对HepG2细胞的抑制作用Fig.6 Inhibition of HepG2 cells by drugs
抑菌圈实验结果见表1和表2。结果表明,络合物C对大肠杆菌,枯草杆菌和金黄色葡萄球菌具有明显的抑菌作用。络合物C对大肠杆菌的MIC值为30 μg/mL、枯草杆菌的MIC值为7.5 μg/mL和金黄色葡萄球菌的MIC值为30 μg/mL。因此,络合物C对枯草杆菌的抑制作用效果最好。
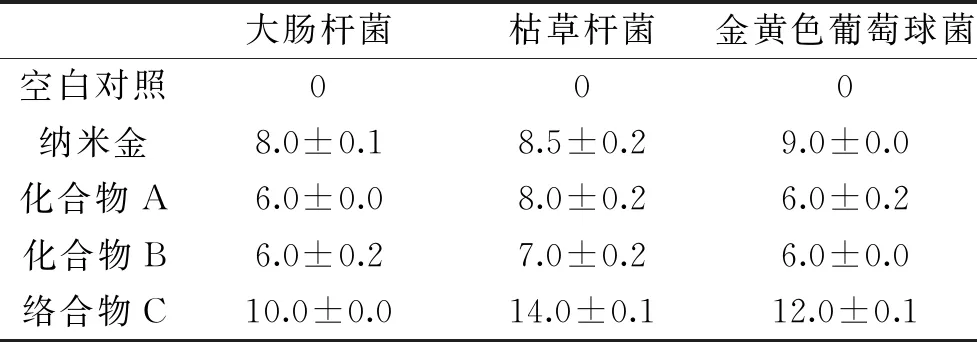
表1 三种细菌的抑菌圈大小(mm)(n=3)Table 1 Inhibition ring size of three kinds of bacterias(mm)(n=3)

表2 络合物C对三种细菌的MICTable 2 MIC of complex C to three kinds of bacterias
3 结论
我国是最早认识和利用虫草的国家。但在虫草素的研究和开发利用方面的水平还比较落后。本实验以化合物A为原料,通过一系列的表征手段证明成功合成化合物B以及络合物C并对其体外活性进行研究。通过MTT实验研究发现二者对HepG2细胞的增殖均有明显的抑制作用,并且发现药物浓度为40~80 μg/mL时,络合物C的抑制作用明显优于化合物B。通过抑菌活性测试结果表明,二者对大肠杆菌、枯草杆菌、金黄色葡萄球菌均有明显的抑菌效果,络合物C对大肠杆菌、枯草杆菌、金黄色葡萄球菌的最小抑菌浓度分别为30、7.5和30 μg/mL。因纳米金颗粒具有良好的生物相容性,使得在人体内可以发挥很好的疗效和作用。纳米药物技术的发展前景可观,其具有巨大的发展潜力和实用价值,本文的研究将为新型创新药物的研究提供一条思路。今后有望在人类疾病治疗领域获得应用。
