10 年随访淋巴瘤样丘疹病1 例
2020-04-30路永红
阳 眉,李 芬,路永红
(1.成都市第二人民医院皮肤科,四川 成都 610017;2.成都市第二人民医院病理科,四川 成都 610017)
1 临床资料
患者,男,47 岁,反复躯干及双下肢红斑、斑块、结节、溃疡10 年,于2016 年6 月29 日来我院就诊。10 年前,患者无明显诱因出现下肢红斑、结节,瘙痒不明显,无疼痛。2007 年出现红斑,且红斑逐渐扩大变硬,形成斑块,能自行消退,但反复发生。结节斑块可破溃结痂,愈合形成大小不一萎缩性瘢痕。患者无乏力、发热、关节疼痛、消瘦及淋巴结肿大。2007年患者曾在“西南医院”皮肤科经皮损活检及免疫组化确诊为“淋巴瘤样丘疹病”,后每6 个月随访1次,偶有新发皮损时,曾使用盐酸米诺环素胶囊0.1,每晚一次,重组人干扰素α-1b 注射液50μg,每周肌注1 次治疗。10 年来无明显自觉症状。个人史无特殊,家族中无类似疾病患者。
系统查体:全身浅表淋巴结无肿大及压痛。专科查体:躯干和四肢可见数十个拇指大斑块,部分表面糜烂,无压痛,大量陈旧性萎缩性瘢痕(图1 ~图3)。口腔和生殖器未累及。

图1 ~图3 躯干和四肢可见数十个拇指大斑块,部分表面糜烂,无压痛,大量陈旧性萎缩性瘢痕
2016 年右大腿皮损组织病理:溃疡边缘鳞状上皮瘤样增生,表皮内中性粒细胞浸润。真皮内散在或簇状分布大小不等的异型细胞,部分异型细胞呈R-S 样。周围伴淋巴细胞、中性粒细胞、嗜酸性粒细胞浸润。皮下组织未见确切异型细胞,(图4 ~图6)。
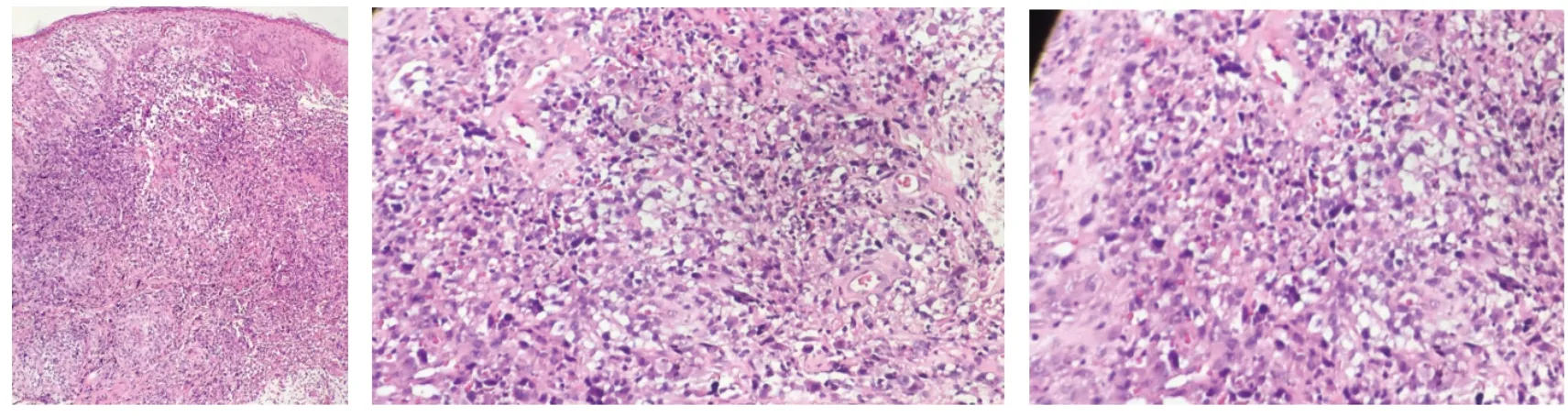
图4 ~图6 HE:表皮增生、表皮与真皮分离,真皮内中性粒细胞、异型淋巴细胞、嗜酸性粒细胞及小淋巴细胞浸润(图4 ×100,图5 ~图6 ×400)
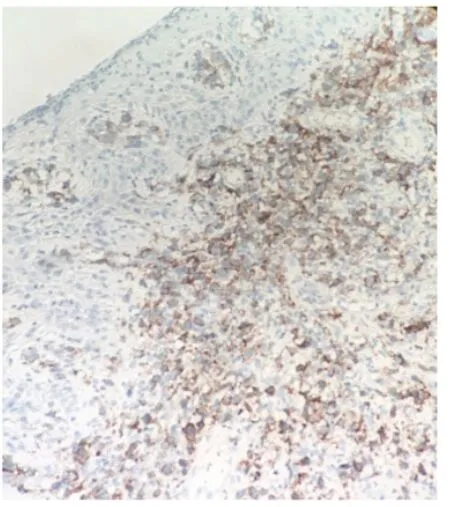
图7 免疫组化:CD30阳性细胞(棕色信号)散在或簇状分布(En-Vision 法×200)
免疫组化:CD30(+,阳性细胞呈簇状或散在小片状分部)、CD3(个别+,背景中部分小淋巴细胞+)、GranzymeB(+)、TIA-1(+);CD20(-)、CD56(-)、ALK(-)、EMA(-)、S100(-)、HMB45(-)、PCK(-);Ki67 阳性率约80%,(图7)。
TCRG 基因重排检测:在目标片段范围内查见克隆性扩增。结合临床表现、组织病理和免疫组化诊断:淋巴瘤样丘疹病(A 型),基因重排检测提示肿瘤细胞有克隆性增生,建议长期随访复查。
本次治疗予以“咪喹莫特乳膏、夫西地酸软膏”外用。电话随访,患者病情稳定,无复发。
2 讨论
淋巴瘤样丘疹病(Lymphomatoid papulosis)是一种慢性复发性、自愈性、丘疹坏死性或丘疹结节性皮肤病,临床上类似急性痘疮样苔癣样糠疹,组织病理表现为CD30阳性的低度恶性淋巴瘤[1]。皮损好发于躯干和四肢,临床表现为红棕色丘疹结节,中央可发生出血、坏死和结痂,(3 ~8)周皮损可自然消退,消退后遗留色素斑或色素沉着斑,偶尔遗留浅表萎缩性瘢痕。病程长短不一,可数月甚至40 余年[1]。诊断需要结合临床、病理、免疫表型,以及必要的长期随访。本病有自限性,预后良好,5 年生存率100%,但有5%~20%患者可发展为其他类型淋巴瘤。不同治疗对病程转归无明确意义,故大多数患者无需特殊治疗,但长期的定期随访十分重要[2]。
本例患者病史10 年余,躯干、四肢皮损反复发作,可自行消退,临床表现和病情发展过程较典型,结合组织病理学特征和免疫组化检查,诊断淋巴瘤样丘疹病(A 型),在长达10 余年的随访观察中,病情稳定,仍宜长期随访。
