细菌表达dsRNA介导桔小实蝇flightin基因的RNA干扰
2020-04-29袁瑞玲郑传伟王艺璇杜春花
袁瑞玲,郑传伟,冯 丹,王艺璇,杜春花,陈 鹏
(1.云南省森林植物培育与开发利用重点实验室,云南 昆明 650201;2.云南省林业和草原科学院, 云南 昆明 650201;3.兴义市林业局,贵州 兴义 562400)
【研究意义】RNA干扰(RNA interference,RNAi)是一种广泛存在于真核生物中高度保守的、由dsRNA诱发的、同源mRNA高效特异性降解,使相应基因不能表达,从而引发基因转录后水平基因沉默的现象[1-3]。dsRNA 导入生物体内后,被细胞中称为Dicer 的RNase Ⅲ分解成21~23 bp的小干扰RNA(siRNA),siRNA 在沉默复合体(RISC)的作用下与目标mRNA 结合,序列特异性地降解靶mRNA,阻止相应蛋白产物的合成,导致靶标基因的功能丧失。【前人研究进展】RNAi技术已被广泛应用在基因功能研究、高通量靶标基因筛选、基因治疗、药物靶标预测等领域,对多种农林害中的成功试验,也证实了利用RNAi进行害虫防治的可能性和广泛性[4-5]。RNAi 技术在害虫防治领域的应用需要大剂量dsRNA,目前最常用的试剂盒合成方法价格昂贵、不能持续生产。利用转基因寄主植物表达昆虫源dsRNA对害虫有一定防效,但植物转基因难度高、研究周期长,具有很大的局限性。用细菌(如大肠杆菌)表达dsRNA的方法操作简单,具有成本低且表达产物量大等特点,是基因工程四大表达系统之一[6]。饲喂或注射细菌表达的dsRNA 能显著干扰甜菜夜蛾[7]、非洲甘薯象鼻虫[8]等害虫的生长,证明该技术在植物保护方面的应用潜力。桔小实蝇[(Bactrocera dorsalis(Hendal)],又名东方果实蝇(oriental fruit fly),属双翅目(Diptera)实蝇科(Trypetidea)、果实蝇属(Bactrocera),是我国二类进境检疫性害虫[9-11],超强的飞行能力使得对桔小实蝇的防治变得困难[12-13]。目前对该害虫的防治手段主要有人工防治、化学防治及性诱剂的生物防治等,这些手段具有一定的防治效果,但总体存在效率低、成本高、对生态环境不友好的问题。因此RNA 干扰这种靶标性强、环境友好型的技术在害虫防控领域展现出较大的应用潜力。
【本研究切入点】飞行蛋白flightin是一种大小在20 ku的多磷酸化肌原纤维蛋白,最早是在黑腹果蝇(Drosophila melanogaster)间接飞行肌中被发现[14]。flightin对昆虫肌肉伸展活化具有重要作用,在间接飞行肌的粗肌丝上与肌球蛋白高度融合,调控和指导粗肌丝的准确组装和收缩等[14]。选择飞行蛋白flightin基因作为RNA干扰靶标,破坏昆虫飞行肌表达通路,阻止昆虫肌肉伸展活化及肌丝的组装和收缩等生理过程,可达到害虫防治的目的。【拟解决的关键问题】本研究以L4440 质粒为载体,构建了利用大肠杆菌HT115 表达桔小实蝇flightin-dsRNA的体系,通过对桔小实蝇新羽化成虫饲喂细菌表达的dsRNA,检测该方法介导的RNAi 在桔小实蝇中的可行性。
1 材料与方法
1.1 试验材料
1.1.1 供试昆虫 供试虫源为云南省林业科学院森保所室内饲养的桔小实蝇种群。饲养条件为:温度25~30 ℃,湿度60%~70%,光周期L16∶D8。
1.1.2 菌株及质粒载体 大肠杆菌DH5α感受态细胞购自天根生化科技(北京)有限公司,大肠杆菌HT115(DE3)购自北京华越洋生物科技有限公司,L4440 质粒购自美国 Addgene 机构,含egfp基因的质粒由西南林业大学徐进研究员惠赠。
1.1.3 酶及有关试剂 总RNA提取试剂盒Trizol-A+、FastKing cDNA第一链合成试剂盒、SYBR PremixEx Taq实时荧光定量PCR试剂盒购自天根生化科技(北京)有限公司,TSINGKE Master Mix DNA Polymerase购自北京擎科新业生物技术有限公司,T4 DNA 连接酶、DNase I、RNaseA、限制性内切酶SacI、XhoI购自thermofisher公司,氨苄青霉素(Amp)、X-gal、IPTG购自Amresco公司,盐酸四环素(Tet)购自solarbio公司,DNA回收试剂盒、质粒小量提取试剂盒购自北京艾德莱生物科技有限公司,异丙醇、氯仿、无水乙醇等其他分析纯试剂购自利安隆博华(天津)医药化学有限公司,引物合成及测序委托北京擎科新业生物技术有限公司完成。
1.2 试验方法
1.2.1 RNA的提取及反转录 桔小实蝇总RNA的提取按照Trizol- A+试剂盒说明书进行。FastKing cDNA第一链合成试剂盒反转录得到cDNA,20℃保存备用,以此作为后续实验 PCR模板。
1.2.2 引物设计 根据桔小实蝇flightin基因转录组序列(GenBank登录号:XM_011210668),用siDirect(http://design.RNAi.jp/)在线预测可能存在的 siRNA 位点,选择其中 678 bp 序列作为RNA 干扰片段。用Primer5.0和Oligo6,结合含有T7聚合酶的L4440干扰载体的多克隆位点以及桔小实蝇flightin基因和egfp序列的限制性内切酶位点设计上游和下游引物。上、下游引物的5'端分别加入SacI和XhoI酶切位点。

1.2.3 桔小实蝇flightin基因及对照egfp基因片段的克隆 分别以桔小实蝇cDNA和含egfp基因的质粒为模板,按照TSINGKE Master Mix DNA Polymerase说明书进行PCR扩增,克隆桔小实蝇flightin及egfp基因干扰片段。PCR扩增体系:TSINGKE Master Mix DNA Polymerase 12.5 μL,上、下游引物和模板各1 μL,加灭菌ddH2O至总体积25 μL。扩增条件:95 ℃预变性3 min;95 ℃30 s、60 ℃ 30 s、72 ℃ 1 min,35个循环;最后72 ℃延伸10 min。反应结束后,用l%琼脂糖凝胶电泳检测并用DNA回收试剂盒纯化回收目的片段送测序。
1.2.4 桔小实蝇flightin及egfp基因的RNA干扰表达载体构建 将L4440质粒与胶回收的桔小实蝇flightin和egfp基因片段分别同时用SacI和XhoI进行双酶切,37 ℃,1 h,分别回收酶切产物。用T4连接酶将回收的L4440载体分别与flightin、egfp基因片段以1∶3比例室温连接10 min。连接产物命名为L4440-flightin、L4440-egfp,分别转化大肠杆菌DH5α感受态细胞。
将转化的感受态细胞涂布在含Amp、IPTG、Xgal的固体LB平板培养基上,37 ℃过夜培养,挑取白色单菌落于含Amp的LB液体培养基中培养,菌液PCR检测阳性后,提取重组质粒L4440-flightin和L4440-egfp,用SacI和XhoI双酶切鉴定,筛选阳性克隆,取阳性克隆菌液送测序。
1.2.5 重组质粒转化大肠杆菌HT115 用热激法将L4440-flightin和L4440-egfp表达载体转入CaCl2法制备的HT115感受态细胞中,涂布于含50 μg/mL Amp和12.5 μg/mL Tet的LB固体培养基上,过夜培养后挑取单菌落接种到Amp和Tet抗性的LB液体培养基中,提取质粒并通过PCR及双酶切鉴定阳性克隆,重组质粒进一步通过测序进行验证。
1.2.6 dsRNA在大肠杆菌HT115中的表达 将带有L4440-flightin和L4440-egfp的HT115菌液接种于Amp和Tet抗性LB液体培养基中,37 ℃振荡培养过夜,1∶25比例将菌液转入新的LB培养基中,37 ℃,200 r/min培养至菌液OD600=0.4左右,加入IPTG终浓度至1.0 mmol/L进行诱导表达,诱导培养4 h后收集菌液。采用TRIzol法提取细菌总RNA,DNase I和RNase A溶液消化纯化总RNA,获得flightin-dsRNA和egfp-dsRNA。
1.2.7 饲喂细菌表达dsRNA的菌液后桔小实蝇flightin基因的RNA干扰 从桔小实蝇羽化开始饲喂IPTG诱导表达靶标dsRNA的大肠杆菌HT115的 10倍浓缩菌液,处理组(饲喂flightindsRNA菌液)和对照组(饲喂egfp-dsRNA菌液)各3个重复,每个重复200头虫(雌雄比1∶1)。将浓缩菌液拌进成虫饲料,每天更换一次新鲜饲料,连续饲喂25 d,处理组和对照组雌、雄虫分开。
RT-qPCR检测RNAi效果:采集1、5、10、15、20日龄虫样品,3次重复,每个重复各取50头虫,分别提取总RNA。荧光定量PCR(RT-qPCR)检测RNAi后flightin基因的表达情况,采用2-△△Ct方法计算靶标基因相对表达量。flightin和egfp基因及16S内参基因的qPCR引物如下:

通过飞行能力测试检测RNAi效果:经过持续饲喂后,分别在5、10、15、20日龄利用佳多飞行磨系统进行雌、雄虫飞行能力测试,参照袁瑞玲等[15]的方法。
整虫与胸部组织质量测定:随机选取待测桔小实蝇各20头,使用乙醚轻微麻醉后迅速称量整虫质量,然后迅速在解剖镜下去除翅、足、头、腹等组织,仅留下胸部肌肉组织快速称量其质量。
试验数据用SPSS19.0统计软件进行方差分析,用Duncan氏新复极差法进行多重比较。
2 结果与分析
2.1 桔小实蝇flightin及对照egfp基因干扰片段的克隆
PCR扩增得到含SacI和XhoI酶切位点的桔小实蝇flightin和egfp基因片段,结果为,桔小实蝇flightin基因片段介于500~750 bp之间,对照egfp基因片段介于100~250 bp之间分别与预期添加了酶切位点和保护碱基后的大小692 bp和201 bp相符(图1 A 3、4泳道)。测序结果与GenBank报道的序列进行比对,无碱基差异,同源性达100%,桔小实蝇flightin基因及egfp基因干扰片段克隆成功。
把载体质粒L4440和胶回收得到的目的片段分别经限制性内切酶SacI和XhoI双酶切后,2 790 bp的L4440质粒线性化单一条带约2 755 bp,桔小实蝇flightin及对照egfp基因大小正确(图1 A 2、5、6泳道)。
2.2 dsRNA表达载体的构建

图1 flightin和egfp基因干扰片段的克隆及干扰载体的构建Fig.1 Cloning of RNAi fragment of flightin and egfp and construction of RNAi vector
HT115菌株中提取的重组质粒L4440-flightin和L4440-egfp(图1 B)经SacI和XhoI双酶切后,1%琼脂糖凝胶电泳20 min检测均分别得到两条带,2 755 bp的线性化L4440质粒载体,及678 bp和187 bp左右的flightin和egfp目的片段(图1 C),表明桔小实蝇flightin基因和对照egfp基因片段成功转入L4440质粒中,与预期设计一致。双向测序结果与目的基因序列完全一致,阅读框正确,说明已经成功构建了L4440-flightin和L4440-egfp干扰表达载体。
2.3 饲喂表达目的dsRNA菌液后flightin表达水平
通过连续饲喂表达flightin-dsRNA和egfpdsRNA的菌液后,处理组与对照组相比,在雌、雄虫中均未产生预期的下调效果,反而出现了不同程度的上调现象。雌虫在饲喂后1、5、10、15、20 d时全部产生上调,与对照相比分别上调1.15、2.70、2.27、1.02、3.50倍,其中,饲喂后5、10、20 d与对照差异显著,饲喂后1、15 d与对照无显著差异。雄虫饲喂后15 d诱发约43%的下调,而在饲喂后1、5、10、20 d分别诱发1.38、2.77、1.57、2.98倍的上调,其中饲喂后5日龄与对照差异显著,其余差异不显著。结果表明,雌、雄虫均在饲喂后5 d诱发的上调幅度最大。
2.4 饲喂表达flightin-dsRNA菌液后对飞行能力的影响
连续饲喂表达flightin-dsRNA和egfp-dsRNA的菌液后,5、10、15、20、25日龄的雌、雄虫飞行能力测试结果(图3)显示,在飞行距离上各日龄处理组与对照组差异不明显。结果表明,饲喂表达flightin-dsRNA的大肠杆菌菌液对桔小实蝇进行RNA干扰,对其飞行能力没有明显影响。
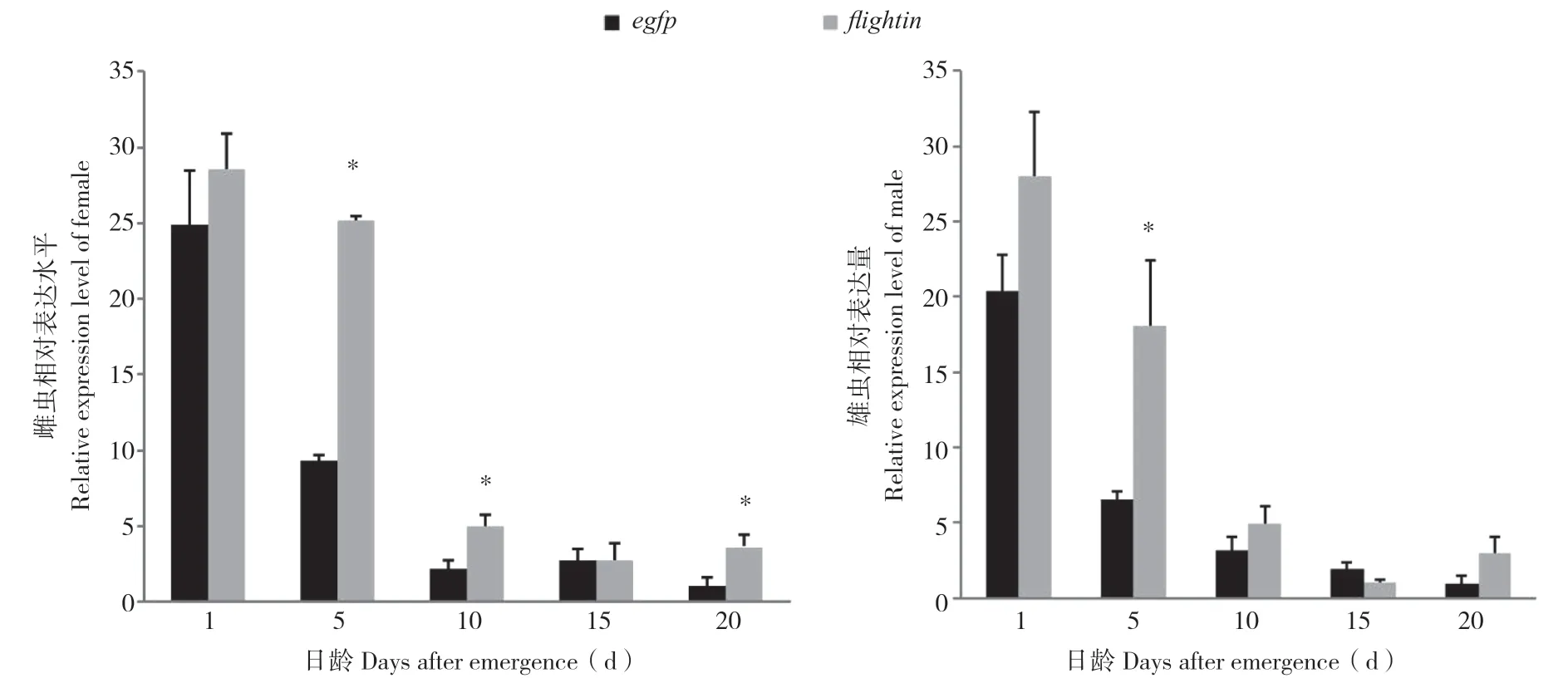
图2 荧光定量PCR分析桔小实蝇flightin基因表达水平Fig.2 Expression level of flightin from Bactrocera dorsalis by real-time quantitative PCR
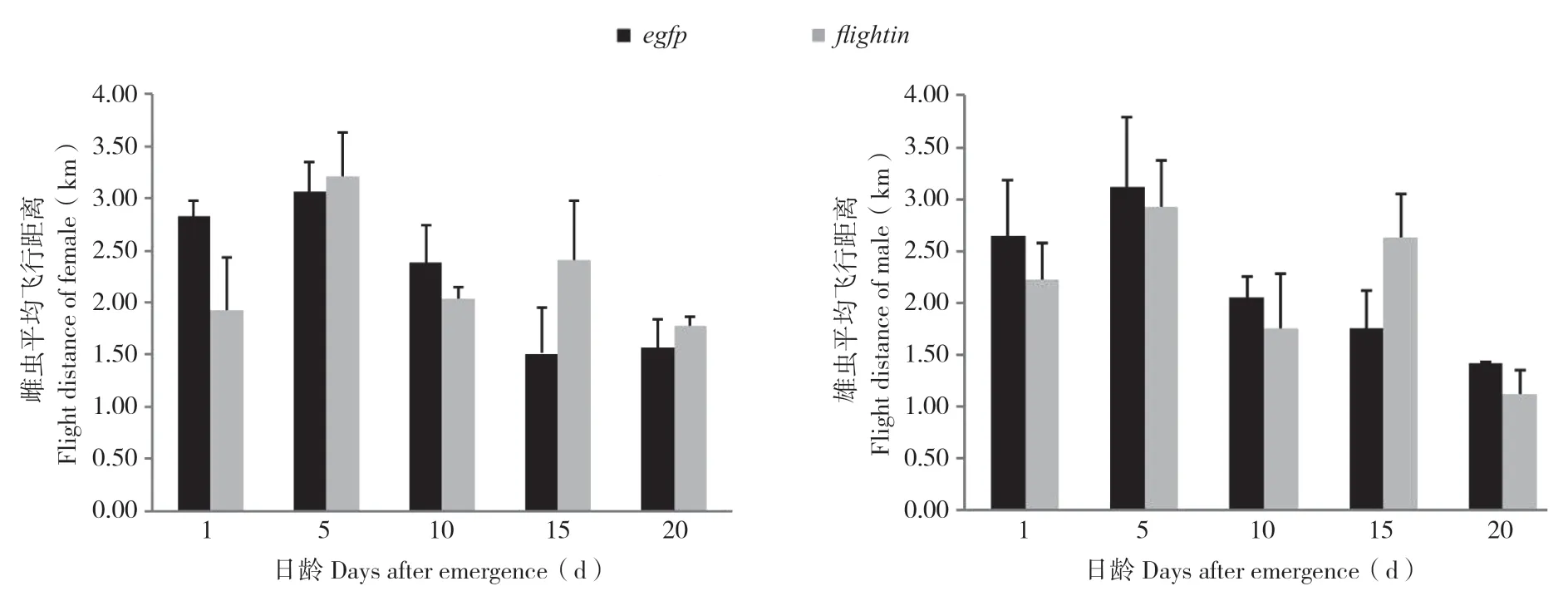
图3 饲喂表达dsRNA菌液后桔小实蝇飞行距离Fig.3 Flight distance of Bactrocera dorsalis after feeding bacterial expressing dsRNAs
2.5 饲喂表达flightin-dsRNA菌液后胸部组织与整虫鲜重比
根据桔小实蝇饲喂菌液进行RNAi后flightin基因的表达量测定结果,选取上调幅度较大的5日龄雌、雄虫称量其整虫与胸部组织的鲜重,得到胸部与整虫的鲜重比。结果显示,处理组雌、雄虫鲜重比分别为34.64%和36.53%,与对照组雌、雄虫鲜重比的34.39%和35.99%没有差异,认为饲喂菌液进行RNAi对胸部肌肉发育无影响。
3 讨论
诸多研究报道,利用大肠杆菌HT115(DE3)工程菌表达dsRNA是经济、高产的方法。早在1998年,Timmons等[16]就开始利用HT115表达的dsRNA喂食线虫,结果靶标基因的表达受到抑制。RNaseIII是细菌中普遍存在的dsRNA 特异性核酸内切酶,由rnc基因编码,HT115 菌株为rnc-突变不能降解自身合成的dsRNA。HT115经过修饰,插入一个λDE3噬菌体衍生物,可通过IPTG诱导表达T7 RNA聚合酶。HT115的四环素和Amp抗性,可在培养过程中分别排除rnc+反突变体和空载质粒。L4440载体序列的多克隆位点两侧各有1个相反方向的T7 聚合酶启动子,转入HT115 菌株后,可经IPTG 诱导表达dsRNA。
随着细菌表达dsRNA技术的不断成熟,该技术在包括桔小实蝇在内的多种昆虫研究中得以实现,有效沉默了目的基因表达[6-7,17-20]。在玉米上喷施表达菌液可抑制花叶病毒的感染[21],在桔小实蝇精子形成相关基因lola、topi、rac、rho、upd和magu研究中,饲喂表达dsRNA的大肠杆菌干扰后,成功降低了基因表达水平并导致精子质量和数量的下降,产卵量及孵化率受干扰而降低[22]。利用该质粒构建的重组质粒可以在大肠杆菌HT115(DE3)中表达大量所需的外源目的dsRNA,在RNAi研究中,该方法比利用试剂盒体外转录和化学合成,能够大大降低实验成本,而且通过诱导表达目的dsRNA饲喂昆虫达到RNAi的操作相对简单易行[6,18]。然而,直接饲喂也存在一些问题,Li等[6]直接喂食桔小实蝇Noa、Rab11、V-ATP-D和Rpl19基因表达dsRNA的大肠杆菌后,成功降低了基因的表达水平,但部分出现上调现象。Liu等[23]研究表明,桔小实蝇成虫长期饲喂dsRNA或表达dsRNA的大肠杆菌菌液,靶标基因均会出现表达上调现象,导致该现象的机制尚不明确。在其他昆虫中不能成功干扰靶标基因的例子也鲜有报道,如黑腹果蝇饲喂表达dsRNA的酵母未能沉默靶标基因表达,对果蝇3种亚种饲喂RNAi体系同样未实现目的基因的沉默等[24-25]。
本研究结果表明,桔小实蝇饲喂表达flightin基因dsRNA的大肠杆菌未造成预期的目的基因转录水平的下调,也未造成飞行能力的减弱或影响发育等,分析认为,未能成功沉默靶标基因的原因可能有:饲喂法对于桔小实蝇存在对RNAi的非基因特异性免疫耐受机制,有关桔小实蝇RNAi免疫耐受研究发现,dsRNA摄取由胞吞机制调控,其间会受到阻遏而无法进入细胞[26];饲喂表达dsRNA大肠杆菌后导致靶标基因上调,存在应激机制及抗性可能是造成这一现象的原因之一[27-28],很多刺激源(包括大肠杆菌)会激发桔小实蝇siRNA通路;Zambon等[29]发现果蝇在抗病毒过程中,出现基因表达上调,结果证实病毒激发了IMD、Toll-way和Jak-STAT的信号通路;不同物种中肠环境差异导致RNAi效率也不尽相同;摄取dsRNA剂量不合适,饲喂法由昆虫自主取食,无法保证其摄取量而影响RNAi效率,也无法确定引发RNAi的dsRNA剂量;桔小实蝇flightin基因在饲喂后1日龄时表达最高,成虫开始饲喂RNAi基本已经过了该基因表达高峰,饲喂法错过最佳干扰时间;有研究表明基于饲喂法的RNAi效率普遍低于注射法[30],本课题组已通过体外转录合成flightin-dsRNA注射桔小实蝇,成功下调了flightin基因的表达,降低了成虫的飞行能力(待发表);也有大量研究表明dsRNA介导的RNAi在双翅目中具有效率不高和持效性差等诸多问题[28]。
4 结论
采用RT-PCR技术克隆桔小实蝇Bactrocera dorsalis(Hendel)flightin基因片段,并以此为靶标,设计有效的干扰片段,以线虫RNAi干扰载体L4440为载体,成功构建了桔小实蝇flightin基因RNAi载体。转化大肠杆菌HT115(DE3)菌株,利用IPTG诱导表达flightin基因对应的dsRNA。通过饲喂表达flightin-dsRNA的大肠杆菌,桔小实蝇flightin基因的表达出现了不同程度的上调;该方法对桔小实蝇飞行能力及胸部肌肉发育未产生明显的影响。未能成功沉默flightin靶标基因的具体原因有待后续研究进一步探明,利用饲喂表达flightin基因dsRNA的大肠杆菌方法对桔小实蝇flightin基因进行RNA干扰的可行性还需进一步确认。
