感染赤星病烟草叶际细菌的多样性分析
2020-04-27汪汉成谢红炼向立刚陈乾丽余知和邹光进
刘 畅,汪汉成,谢红炼,向立刚,黄 宇,陈乾丽,余知和,邹光进
1. 长江大学生命科学学院,湖北省荆州市荆州区荆秘路88 号 434025
2. 贵州省烟草科学研究院,贵阳市观山湖区龙滩坝路29 号 550081
3. 长江大学农学院,湖北省荆州市荆州区荆秘路88 号 434025
4. 贵州大学农学院,贵阳市花溪区花溪大道南段2708 号 550081
5. 贵州烟草公司黔东南州公司,贵州省黔东南苗族侗族自治州凯里市韶山南路31 号 556000
在烟草生长过程中易被多种植物病原菌侵染,进而造成烟草田间病害的发生[1]。据统计,未使用药剂防治时烟草因各种叶部病害可引起较大的经济损失[2]。近年来,贵州省部分烟区烟草赤星病频发,且造成的损失较大[3]。赤星病菌是引起烟草生长发育中后期病害的主要病原菌,病原菌侵入所导致的叶际微生物种群结构变化通常会造成相关病害的发生,赤星病菌侵染后,是否有其他附生微生物参与危害目前尚不清楚。但烟草叶部病害的发生与叶际微生物群落结构及其生态环境密切相关。高爽等[4]综述了叶际微生物群落的组成、特点及其与外界环境交互作用,发现已报道的叶际微生物群落组成均存在一定规律,表明微生物群落并不是随机组成[5-9]。Finkel 等[8]采集了以色列及美国不同地区的树叶样品进行相关微生物群落结构组成分析,发现地理位置决定了排盐荒漠树系统球体微生物群落的种群结构,不同种类叶际微生物群落的Tamarix 属高度相似,而生长在不同气候区的同一物种的树木存在不同的微生物群落。其中,细菌是叶际微生物中数量最多的一种[10]。随着高通量测序技术的发展,基于扩增子测序的分析技术被用于叶际微生物多样性的研究[11]。Lindow 等[12]研究发现,叶际微生物有引发植物病害的作用;孙泓等[13]研究了不同生境中桂花和夹竹桃叶际细菌的群落结构,提出植物种类、生态环境及二者的交互作用均能显著影响叶际细菌群落结构。在烟草上,徐慧等[14]采用传统分离鉴定方法测定了健康烟叶上的细菌分布,发现不同产地烟草叶片上细菌种类不同,分离得到19 个属共129 株细菌。该试验结果有助于了解烟叶叶际及内生菌组成。但传统分离鉴定方法存在一定局限性,无法对不可培养微生物进行检测[15],也无法测定其在群落中的相对含量。同时是否有叶际附生细菌参与了赤星病的侵染与危害,目前尚不清楚。为此,采用IonS5XL 高通量测序技术分析了感染赤星病烟草叶际附生细菌群落结构与多样性,旨在明确与赤星病发生相关的烟草叶际细菌种群结构,为烟草赤星病的有效防治提供依据。
1 材料与方法
1.1 试验基本情况及样品采集
黔东南州烟区(26°36’N,107°59’E)属中亚热带季风湿润气候区,具有冬无严寒,夏无酷暑,雨热同季的特点。年平均气温14~18 ℃。境内年日照时数为1 068~1 296 h,无霜期270~330 d,降雨量1 000~1 500 mm,相对湿度为78%~84%。供试品种云烟87,种子消毒后采用漂浮育苗,烟苗长至8 片真叶期时移栽,按当地优质烟叶生产技术规范进行田间管理。2018 年8 月29 日,在贵州省烟草赤星病爆发严重的烟田进行样品采集。田间随机选取3 株感赤星病严重的烟株,剪刀经酒精消毒后分别剪取每烟株发病叶片样品,置于无菌取样袋中并分别编号。将样品放入低温保藏箱,并迅速带回实验室,置于-80 ℃的冰箱中保存、备用。
1.2 试剂与仪器
试剂盒及测序系统:GeneJET 胶回收试剂盒和Ion Plus Fragment Library Kit 48 rxns 建库试剂盒(美国Thermofisher 公司)。IonS5XL 测序系统由北京诺禾致源科技股份有限公司提供。
1.3 方法
1.3.1 样品DNA 提取、扩增及纯化
采用CTAB 法对样品基因组DNA 进行提取,取5 μL 提取物于离心管中,用无菌水稀释至1 ng/μL,检测样品DNA 的纯度和浓度。
样品16S 区的PCR 扩增以稀释后的样品基因组DNA 为 模 板,采 用 引 物515F(5’-GTGYCAG CMGCCGCGGTAA-3’)和806R(5’-GGACTACH VGGGTWTCTAAT-3’)进行扩增。PCR 扩增体系为30 μL:2×Phusion High-Fidelity PCR Master Mix with GC Buffer 15 μL,引物(2 μM)3 μL,gDNA(1 ng/μL)2 μL,用灭菌的ddH2O 补至30 μL。产物纯化后回收目标条带。使用GeneJET 胶回收试剂盒回收产物。
1.3.2 16S 文库构建及高通量测序
使用Ion Plus Fragment Library Kit 48 rxns 建库试剂盒构建文库,采用Qubit 定量。文库检测合格后,使用IonS5XL 测序系统进行上机测序。以上步骤均由北京诺禾致源科技股份有限公司完成。
1.3.3 数据分析
将IonS5XL 下机数据导出fastq 文件,根据barcode 序列区分各个样品的数据,barcode 允许的错配数为0,最大引物错配数为2。采用Cutadapt(V1.9.1, http://cutadapt.readthedocs.io/en/stable/)[16]对数据进行剪切拆分等相关质控,Reads 序列通过https://github.com/torognes/vsearch/[17]与物种注释数据库进行比对,去除嵌合体序列[18],得到最终的有效数据(Clean Reads)。
利用Uparse 软件(Uparse v7.0.1001,http://www.drive5.com/uparse/)[19]对 全 部Clean Reads 进 行 聚类,默认以97%的一致性(Identity)将序列聚类成为可操作分类单元OTUs(Operational Taxonomic Units),并进行物种注释。使用Mothur 方法与SILVA132(http://www.arb-silva.de/)[20]的SSUrRNA数据库[21](设定阈值为0.8~1)获得相关分类学信息,并分别在各个分类水平上统计群落组成。快速 多 序列 比 对 使 用MUSCLE[22](Version 3.8.31,http://www.drive5.com/muscle/)软件进行,以得到所有OTUs 序列的系统发生关系。然后以样品中数据量最少的为标准,对各样品的数据进行均一化处理。使用Qiime 软件(Version 1.9.1)计算各Alpha 多样性指数,使用R 软件(Version 2.15.3)绘制稀释曲线。并以分析为基础进行基于OTUs、物种组成的聚类分析和统计比较,挖掘样品之间的物种组成差异。
2 结果与分析
2.1 16S 序列测序深度分析及数据质控
图1 稀释曲线(Rarefaction curve)展示了不同样品间多样性的差异。本次测序3 个样品的稀释曲线在测序深度为30 000 时趋于平缓,表明此时增加测序数据无法再找到更多的OTU。因此,可以认为测序数据量足以反映样品中的物种多样性,测序深度已经覆盖到样品中的所有物种。

图1 稀释曲线(OTU 水平)Fig.1 Rarefaction curve(OTU level)
共获得3 个感赤星病烟草叶际样品,分别为YBBqdL1x、YBBqdL2x、YBBqdL3x。原始序列经优化处理后,3 个样品共得到24 0176 条高质量序列片段,97 919 144 个碱基,单一样品序列数在80 047~80 074 条之间,序列平均长度为407 bp,共检测到78 个OTUs。
2.2 OTU 聚类分析
在97%的相似度水平下对样品序列进行OTU聚类,3 个叶片样品共鉴定出细菌的5 个门,8 个纲,17 个目,22 个科,25 个属,18 个种,66 个OTU,见表1。
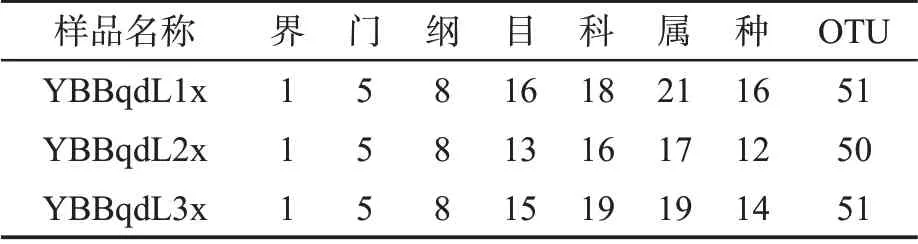
表1 感染赤星病烟草叶际细菌群落不同分类水平的总量Tab.1 Total amount of bacterial communities in different tobacco plants infected by brown spot disease at different taxonomic levels (个)
Venn 图分析结果(图2)表明,在OTU 水平下,3 个样品之间细菌种类较为接近,共有的OTU 种类为37 个,远高于各样品中独有的种类。

图2 感染赤星病烟草叶际细菌群落Venn 图Fig.2 Venn diagram illustrating the numbers of unique and shared OTUs of bacterial community of brown spot disease
2.3 微生物多样性指数分析
基于OTU 水平的Alpha 多样性指数(表2)表明,YBBqdL1x 与YBBqdL3x 样品检测到的细菌OTU 数量略高于YBBqdL2x,但无显著差异。3 个样品的覆盖度指数(Coverage index)均到达了0.999 以上,表明测序结果合理。丰富度指数(Richness index)Chao1、Ace 均为YBBqdL1x 样品细菌群落丰富度高于YBBqdL2x 与YBBqdL3x。多 样 性 指 数(Diversity index)Shannon 指 数 及Simpson 指数中,均为YBBqdL3x 样品多样性高于YBBqdL1x 与YBBqdL2x;PD_whole_tree 指 数 中,YBBqdL1x 样品高于YBBqdL2x 与YBBqdL3x。

表2 感染赤星病烟草叶际细菌群落Alpha 多样性指数分析(OTU 水平)Tab.2 Alpha diversity index of phyllosphere bacterial community in tobacco leaves infected by brown spot disease(OTU level)
2.4 细菌群落组成与结构分析
在门水平上,YBBqdL1x 的群落主要组成为变形菌门Proteobacteria(85.35%)、厚壁菌门Firmicutes(1.06%)、放线菌门Actinobacteria(0.30%);YBBqdL2x的群落主要组成为变形菌门Proteobacteria(79.45%)、厚壁菌门Firmicutes(0.65%)、放线菌门Actinobacteria(0.02%);YBBqdL3x 的群落主要组成为变形菌门Proteobacteria(49.97%)、厚壁菌门Firmicutes(0.72%)、放线菌门Actinobacteria(0.47%),见图3。
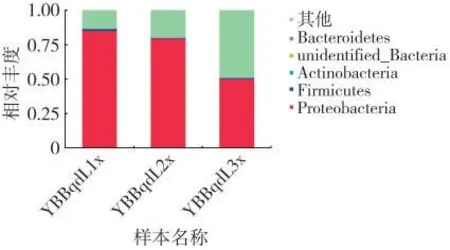
图3 不同样品细菌群落门水平的相对丰度Fig.3 Relative abundances of bacterial community phyla in different samples
在属水平上,YBBqdL1x 的群落主要组成为泛菌属Pantoea(40.88%)、假单胞菌属Pseudomonas(25.54%)、鞘氨醇单胞菌属Sphingomonas(8.58%)、甲醇杆菌属Methylobacterium(7.65%)、Aureimonas(1.58%)、Falsirhodobacter(0.05%)、细 杆 菌 属Microbacterium(0.03%)、乳 酸 菌 属 Lactobacillus(0.12%)、Fusicatenibacter(0.21%)、Blautia(0.19%)、双歧 杆 菌 属Bifidobacterium(0.15%)、粪 杆 菌 属Faecalibacterium(0.15%)、四折叠球菌属Quadrisphaera(0.12%)、毛螺旋菌属Anaerostipes(0.09%)、Romboutsia(0.03%);YBBqdL2x的群落主要组成为泛菌属Pantoea(40.15%)、假单胞菌属Pseudomonas(33.59%)、鞘氨醇单胞菌属Sphingomonas(1.36%)、甲醇杆菌属 Methylobacterium(1.24%)、Falsirhodobacter(0.47%)、Aureimonas(0.13%)、细杆菌属Microbacterium(0.02%)、乳 酸 菌 属 Lactobacillus(0.23%)、粪 杆 菌 属Faecalibacterium(0.02%)、肠 球 菌 属Enterococcus(0.02%)、Candidatus_Saccharimonas(0.03%)、螺杆菌属Helicobacter(0.03%);YBBqdL3x 的群落主要组成为 泛 菌 属 Pantoea(12.66%)、甲 醇 杆 菌 属Methylobacterium(9.63%)、假单胞菌属Pseudomonas(8.20%)、鞘氨醇单胞菌属Sphingomonas(8.14%)、Aureimonas(4.55%)、Falsirhodobacter(3.55%)、细杆菌属Microbacterium(0.35%)、Novosphingobium(0.31%)、乳 酸 菌 属Lactobacillus(0.23%)、纤 维杆 菌 属Cellulomonas(0.12%)、肠球菌属Enterococcus(0.11%)、黄单胞杆菌属Xanthomonas(0.07%)、Roseomonas(0.05%)、螺杆菌属Helicobacter(0.02%),见图4。

图4 不同样品细菌群落属水平的相对丰度Fig.4 Relative abundances of bacterial community genera in different samples
属水平的相对丰度热图通过颜色的变化直观显示了各样品在属水平相对丰度位列前30 的群落分布情况,见图5。
使用UPGMA(Unweighted Pair Group Method with Arithmetic mean)算术平均非加权配对群聚类方法,输入Bray Curtis 距离矩阵,将各样品在门水平上的物种相对丰度进行聚类,结果见图6。通过颜色变化直观显示出不同样品在门水平上相对丰度位列前10 的分布情况,热图左侧的UPGMA 聚类树表明:3 个样品细菌群落结构存在差异,因而聚集为两大类。其中YBBqdL2 和YBBqdL3 样品群落结构较相似可聚集为一类,YBBqdL1 样品聚为一类,但3 个样品总体差异不大。
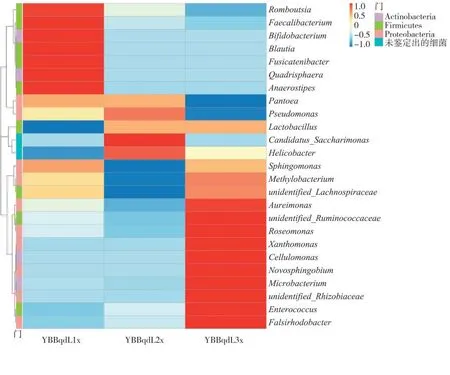
图5 不同样品细菌群落属水平的相对丰度热图Fig.5 Heat map of relative abundances of bacterial community genera in different samples

图6 基于Unweighted Unifrac 距离的非加权组平均法聚类分析Fig.6 UPGMA clustering tree based on Unweighted Unifrac distance
3 讨论
感染赤星病烟草叶片3 个样品扩增的16S rRNA 基因的V3~V4 区域的长度均为407 bp,与实际V3~V4 区域的长度基本吻合,结果证实了IonS5XL 高通量测序技术研究感赤星病烟叶叶际细菌群落结构的可行性。
与建立在纯种分离和纯培养技术基础之上的传统研究方法相比,在对叶际微生物总群落结构分析上,高通量测序技术可鉴定出部分不可培养菌种以及更多种类可培养细菌。IonS5XL 系统采用半导体测序技术,与其他基于光学原理的测序仪不同,无需使用复杂的光学元件或带标记的核苷酸,在保证质量的同时可达到较高的测序速度。
叶际微生物和宿主植物、叶际微生物之间都会发生复杂的交互作用[23],微生物和宿主之间包括寄生、共生和互利共生的关系。本试验中对细菌群落结构分析发现,泛菌属Pantoea(31.23%)、假单胞菌属Pseudomonas(22.44%)、甲醇杆菌属Methylobacterium(6.17%)、鞘 氨 醇 单 胞 菌 属Sphingomonas (6.02%)、 Aureimonas (2.08%)、Falsirhodobacter(1.35%)为感赤星病烟叶叶际中优势菌属(组成所占比例1%以上),此结果与徐慧等[14]测定的烟草叶际细菌多样性结果基本一致,多数叶际定殖的细菌推测可能与烟叶多为共生关系。群落组成占比最高的泛菌属Pantoea 与烟草赤星病病原菌Alternaria alternata 之间是否存在一定的关系还有待进一步深入研究。在烟草赤星病病原菌侵染烟草过程中,其在叶际微生物环境中占主导地位,叶际细菌群落结构尚未发生明显的变化,推测烟草赤星病菌与大部分叶际细菌可共生,同时可导致环境中腐生菌在烟草叶际的富集。烟叶生产上烟草赤星病防治多为使用化学杀菌剂,已有学者进行了大量相关研究[24],而从增加生防细菌含量方面来有效防治烟草赤星病有待于进一步试验。
对感赤星病烟草叶际微生物进行测序分析,有利于揭示烟草赤星病叶际细菌群落结构及多样性。通过群落组成分析,各细菌群落在烟草赤星病发生中后期叶际间的作用以及各细菌群落间的关系则需要进一步深入研究。另外本试验中测序样品数量有限,且仅局限于贵州省黔东南州烟区,因此样品数量还有待增加,采样范围有待扩大,同时还应增加不同感病程度的样品,以分析在烟草赤星病发生过程中细菌群落结构的变化规律性。
4 结论
感染赤星病烟草叶际细菌群落优势菌属为泛菌属Pantoea(31.23%)、假单胞菌属Pseudomonas(22.44%)、甲醇杆菌属Methylobacterium(6.17%)、鞘氨醇单胞菌属Sphingomonas(6.02%)、Aureimonas(2.08%)和Falsirhodobacter(1.35%)。烟草赤星病菌可与大部分叶际细菌共生,同时烟草赤星病的发生或可导致环境中腐生菌在烟草叶际的富集。烟草赤星病病原菌链格孢菌的侵染没有明显改变叶际细菌群落结构。
