固相萃取—微波衍生化—GC-MS法同时测定水中6种雌激素物质
2020-04-25袁宏宇王欣泽迟莉娜字建婷王荣会
袁宏宇,王欣泽,,沈 剑,迟莉娜,字建婷,王荣会
(1. 上海交通大学 环境科学与工程学院,上海 201100;2. 上海交通大学 云南(大理)研究院,云南 大理 671000)
环境雌激素(Environmental estrogens,EEs)是属于环境内分泌干扰物的一类化学物质,进入生物体会破坏机体稳定性和正常调控作用[1],包括天然内源雌激素和人工合成外源雌激素两大类。天然内源雌激素通过动物及人体排泄物释放到环境中,包括雌酮(E1)、17β-雌二醇(β-E2)、17α-雌二醇(α-E2)、雌三醇(E3)[2]等。化学工业技术的飞速发展导致大量合成有机化学品释放到环境中,其中17α-乙炔基雌二醇(EE2)、双酚A(BPA)是具有内分泌干扰效应的典型雌激素类物质[3-5]。大量研究表明,雌激素类物质在浓度极低(ng/L级别)情况下就能对生物体的内分泌系统等造成损伤,且双酚A、EE2与天然雌激素之间可能还会存在协同效应[6-7]。
气相色谱-质谱联用方法(GC-MS)因其分离效果好、灵敏度高及成本相对较低等优点成为目前最为常用的EEs分析测定方法[8-10],由于EEs具有痕量特征,通常需要经过过滤、富集及浓缩等复杂前处理步骤,从而给分析测定带来了极大困难。目前常用的前处理方法包括固相萃取(SPE)[11-12]、液液萃取(LLE)[13-14]、固相微萃取(SPME)[15]等,其中固相萃取因其简便高效等优点得到广泛应用。
本工作系统优化了气相色谱柱升温程序、衍生化条件以及固相萃取小柱类型,建立了快速同时测定水中这6种雌激素物质的分析方法,对研究水体环境雌激素内分泌干扰效应具有重要应用价值。
1 实验部分
1.1 仪器和设备
TRACE1300型GC,ISQ Series Quadrupole 型MS气相色谱质谱联用仪:美国ThermoFisher公司;TriPlus RSH型自动进样器:美国ThermoFisher公司;SQP型电子天平:德国赛多利斯公司;SBAB-57044型CNW 12位固相萃取装置:上海安谱实验科技股份有限公司;HC-C18型、Poly-Sery HLB型、Poly-Sery PSD型、ENVI-18 SPE型等4种萃取小柱:上海安谱实验科技股份有限公司。
1.2 试剂及标准溶液配制
雌激素分析标准物(E1、17α-E2、17β-E2、E3、EE2、BPA)、标准替代物(17β-E2-2,4-d2)以及内标物(灭蚁灵):均为分析标准品,均购自阿拉丁试剂有限公司;含有1% (w,下同)三甲基氯硅烷(TMCS)的双(三甲基硅烷基)三氟乙酰胺(BSTFA):购自上海麦克林公司。
甲醇:色谱级,纯度≥99.9%;乙酸乙酯:HPLC级,纯度≥98.0%;正己烷:HPLC级,纯度≥98.0%;无水吡啶溶液:纯度≥99.8%。
精确称量6种目标物质的标准品以及标准替代物各50 mg,分别溶于50 mL甲醇中,配制成1 g/L的高浓度单标溶液,分别移取100 μL到10 mL容量瓶中,再加入甲醇配制成10 mg/L的混合标准溶液。准确称量50 mg 灭蚁灵溶于50 mL 正己烷中,配制成1 g/L 的高浓度内标溶液。配制好的溶液放置于-20 ℃冰箱中冷冻避光保存,根据需要逐级稀释使用。
1.3 实验方法
1.3.1 固相萃取
取200 μL各雌激素质量浓度分别为1 mg/L的混合标准溶液加入到1 L超纯水中,配成雌激素质量浓度分别为200 ng/L的水样。选择HC-C18、Poly-Sery HLB、Poly-Sery PSD以及ENVI-18等4种不同类型的萃取小柱对水样进行固相萃取,考察不同萃取小柱对6种雌激素物质的回收率。每组实验重复3次,计算平均回收率及标准偏差。
固相萃取分为以下4个步骤:1)萃取小柱活化与平衡,先分别取5 mL乙酸乙酯与5 mL甲醇过柱,使萃取小柱活化,再用5 mL超纯水清洗2次,流量约为2 mL/min;2)水样过柱,用真空泵抽取过滤水样通过萃取小柱,富集待测物于填料中;3)淋洗杂质,用10 mL 10%(w)甲醇溶液洗涤小柱,抽真空30 min左右直至小柱表面干裂;4)洗脱目标物,用10 mL乙酸乙酯洗脱萃取小柱中的待测物,控制流量为2 mL/min左右,并用试管收集洗脱液。
1.3.2 衍生化
将固相萃取洗脱液(衍生化优化实验时以配制的雌激素混合标准溶液代替固相萃取洗脱液)在30 ℃条件下通入温和高纯氮气,干燥完全后加入25 μL(BSTFA+1% TMCS)+50 μL吡啶作为衍生化试剂,一定微波功率下加热一定时间,再在30 ℃条件下用温和氮气干燥,加浓度为1 mg/L的内标物灭蚁灵溶液定容至400 μL,转入2 mL进样瓶,待GC-MS分析。
1.3.3 GC-MS
1)GC条件:TR5-MS型石英毛细管色谱柱(30 m×0.25 mm×0.25 μm);99.999%高纯氦气为载气,采用恒流模式(载气流量1.2 mL/min);不分流进样,每次取样量为1.0 μL;隔垫吹扫5.0 mL/min。实验考察色谱柱升温程序。
2)MS条件:电子轰击离子源(EI),电离电压70 eV;离子源温度250 ℃,传输线温度280 ℃;溶剂延迟时间10 min。采用全扫描模式进行定性分析、离子扫描模式进行定量测定,全扫描质量数范围(m/z)为33~650。使用Xcalibur软件进行定性分析,确定6种雌激素目标物、标准替代物及内标物所对应的标准谱图以及保留时间,选择3~4个相对丰度较强、分子量较大的碎片离子作为定性离子,并选择其中丰度最强且与其他物质特征离子没有干扰的定性离子作为定量离子。
1.3.4 标准曲线、方法检出限及线性范围的确定
用含有6种目标物的混合标准溶液配制成各物质含量分别为1,3,5,10,25,50,100,200,300 ng的梯度浓度系列标准溶液,再分别加入物质含量为100 ng的17β-E2-2,4-d2(标准替代物)溶液,在优化条件下进行固相萃取—微波衍生化—GC-MS分析测定,以各目标物的定量离子峰与内标物的定量离子峰的峰面积比值为纵坐标Y,以各目标物质量浓度X(μg/L)为横坐标进行线性回归分析。以信噪比S/N =3结合目标物回收率计算分析方法的检出限(LOD),以信噪比S/N=10计算该方法的定量限(LOQ)。
2 结果与讨论
2.1 色谱柱升温程序的选择
气相柱温箱的升温程序影响着目标物的保留时间、峰分离度以及峰形等,对定性与定量分析均会产生一定的影响。设置3种不同的升温程序(见表1),考察全扫描色谱图出峰效果。实验结果表明,程序升温方式2条件下雌激素物质的全扫描总离子色谱谱图中6种标准物质及内标物均出峰、峰形良好且分离度较好,见图1。因此确定柱温箱最佳升温程序为:初始温度50 ℃,保持2 min;以20 ℃/min的升温速率上升至260 ℃,保持5 min;再以3 ℃/min的升温速率上升至280 ℃,保持5 min。在此升温程序条件下,总扫描时长为24.62 min。

表1 3种程序升温方式
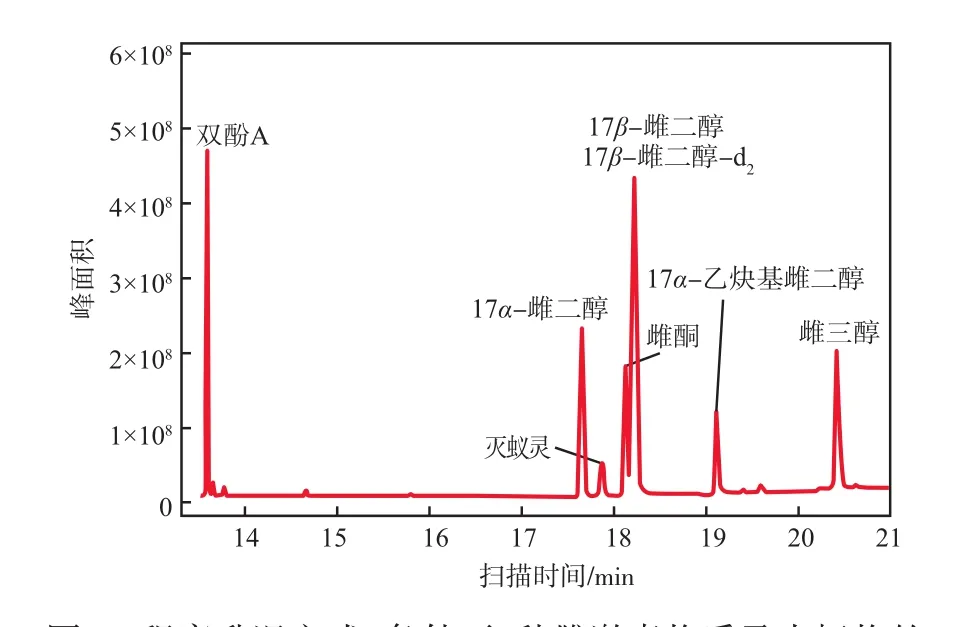
图1 程序升温方式2条件下6种雌激素物质及内标物的全扫描总离子色谱谱图
2.2 色谱与质谱特征
根据图1数据进行定性分析,得到每种雌激素物质和内标物的保留时间及特征离子,见表2。
2.3 衍生化条件优化
由于雌激素类物质为弱极性至中等极性化学物,毛细管色谱柱为非极性柱,目标物质直接进样的出峰效果差,且会对色谱柱造成损害,故采用BSTFA+1%TMCS+吡啶作为衍生化试剂。目标物经衍生化后,产物单一稳定,分离效果好,灵敏度高[16-17]。
2.3.1 衍生化加热方式的选择
因可通过提高极性分子运动速度和碰撞频率快速加热物质、快速提高物质之间的化学反应速率,微波在样品前处理中得到了广泛应用[18-21]。为了提高衍生化效率,本实验将微波加热衍生化与水浴及烘箱加热衍生化的效果进行了对比研究。预实验结果表明:水浴及烘箱加热衍生化的最佳条件为40 ℃反应20 min,这与王园园等[22]的研究结果一致,加热温度过高可能会生成不利于GC-MS分析的副产物,延长反应时间,对提高目标物回收率贡献不大。衍生化方式对雌激素物质峰面积的影响见图2。由图2可见:常温20 ℃条件下峰面积最小,说明衍生化效果最差;水浴40 ℃和烘箱40 ℃加热条件下峰面积大致相当;微波252W加热3 min衍生化的各目标物峰面积略低于水浴和烘箱加热;微波567 W加热3 min的衍生化效果最佳,各目标物的峰面积相比常温20℃衍生化增加了24.09%~42.97%,相比烘箱加热衍生化增加了12.12%~24.31%,相比水浴加热衍生化增加了8.06%~33.53%,相比微波252 W加热3 min衍生化增加了16.62%~28.12%。微波加热使衍生化时间大幅缩短至3 min,与传统衍生化方式相比,衍生化效率显著提升。
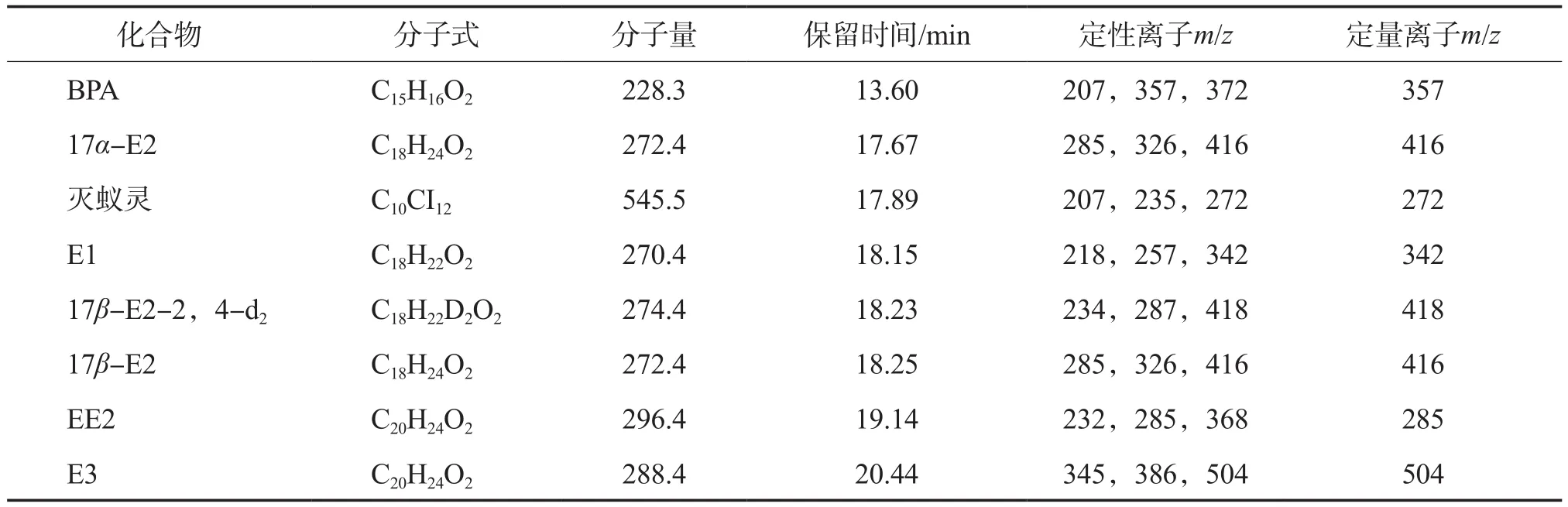
表2 雌激素物质和内标物的保留时间及其特征离子
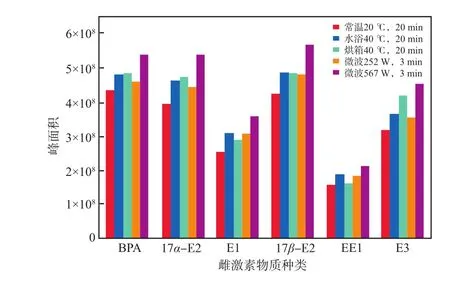
图2 衍生化方式雌激素物质的峰面积
2.3.2 微波衍生化条件
微波功率和加热时间是影响微波衍生化效果的关键因素[23-24]。采用BSTFA +1%TMCS+吡啶作为衍生化试剂,分别考察不同微波功率和不同加热时间对6种雌激素物质衍生化效果的影响,每组实验条件重复3 次,计算平均峰面积。
2.3.2.1 微波功率
在微波加热时间为3 min的条件下,微波功率对雌激素物质峰面积的影响见图3。由图3可见:252 W微波加热功率下,雌激素物质峰面积均较小,说明衍生化反应不完全导致衍生化效果差;微波功率升高至315 W时,雌激素物质峰面积均达到最大,说明衍生化效率达到最佳;微波功率调至406 W时雌激素物质峰面积明显减小,再继续提高微波功率也无明显变化,说明微波功率过高时部分衍生化产物可能发生分解或与衍生化试剂进一步反应生成了不利于GC-MS分析的副产物,导致雌激素物质峰面积减小,衍生化效果变差。
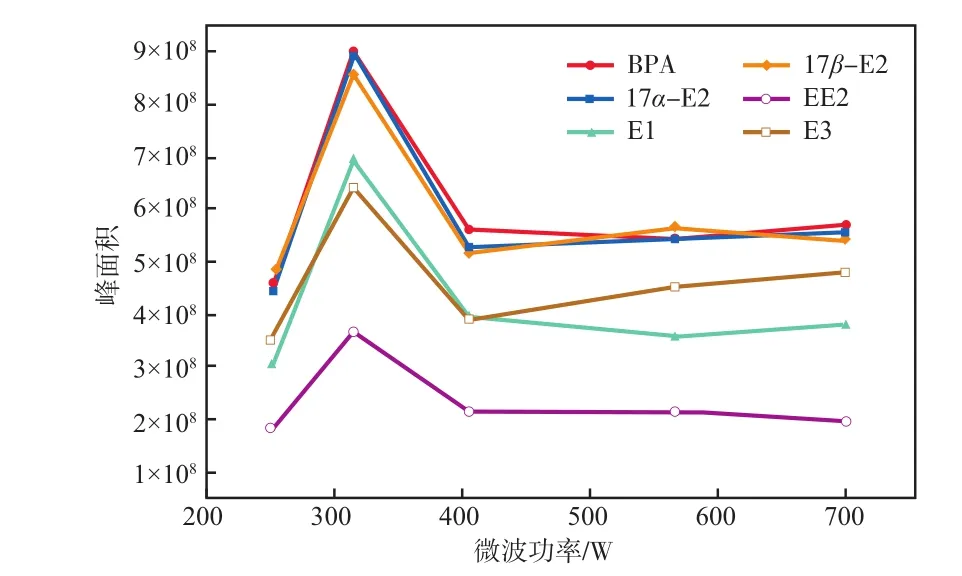
图3 微波功率对雌激素物质峰面积的影响
2.3.2.2 微波加热时间
在微波功率315 W条件下,微波加热时间对雌激素物质峰面积的影响见图4。
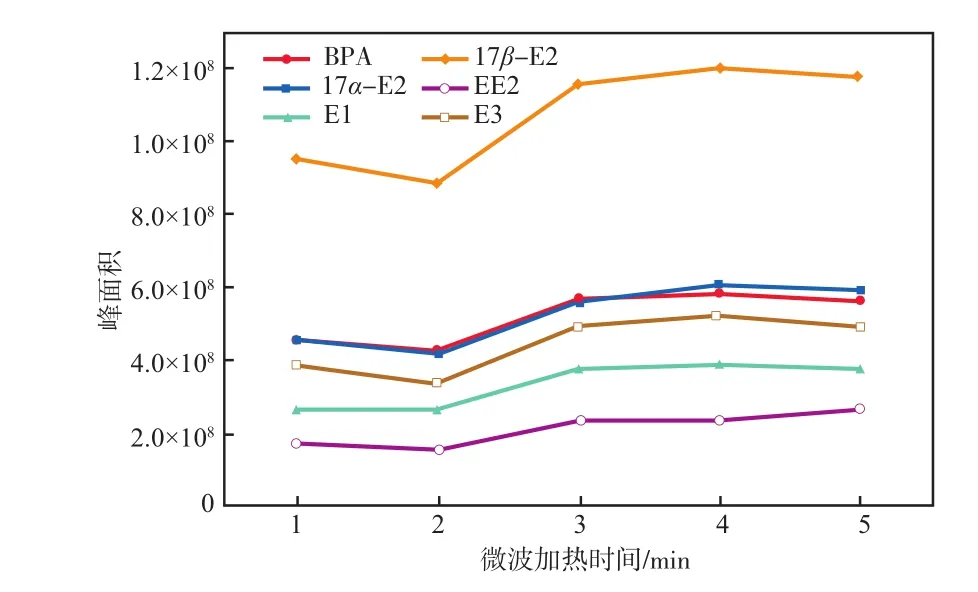
图4 微波加热时间对雌激素物质峰面积的影响
由图4可见:随着微波加热时间增加,雌激素物质峰面积逐渐增大;当微波加热时间为4 min时,6种雌激素物质的峰面积均最大,衍生化效果最好;再增加微波加热时间则衍生化效果无明显变化。为了防止衍生化产物分解并保证最佳衍生化效率,所以选择微波功率315 W条件下加热4 min为最佳衍生化条件。
2.4 固相萃取条件优化
4种不同类型萃取小柱吸附6种雌激素物质的回收率见图5。
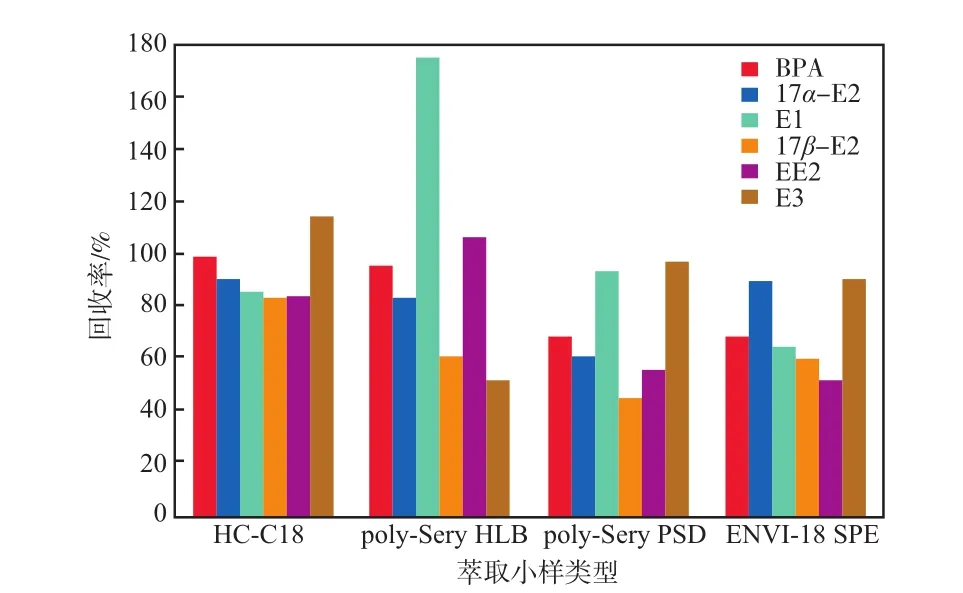
图5 4种不同类型萃取小柱吸附6种雌激素物质的回收率
由图5可见:HC-C18小柱对6种雌激素物质的回收率范围为82.93%~115.00%,对6种雌激物质吸附效果良好;Poly-Sery HLB小柱对6种雌激素物质的回收率范围为50.95%~174.14%,回收率波动范围偏大,其中对E3和17β-E2吸附效果较差,回收率分别只有50.95%和60.51%;Poly-Sery PSD小柱对6种雌激素物质的回收率范围为45.15%~93.10%,对E1和E3的回收率均高于90%,但对其余4种目标物的回收效果相对较差,均低于70%;ENVI-18 SPE小柱对6种雌激素物质的回收率范围为52.21%~90.18%,对E3和17α-E2吸附效果较好,其余目标物则效果一般。综上所述,HC-18小柱对6种雌激素物质的吸附效果比其他3种小柱好,且吸附稳定。
2.5 标准曲线、方法检出限及线性范围
6种雌激素物质的线性回归方程、相关系数、检出限、定量限及线性范围见表3。由表3可见,各目标物峰面积与其质量浓度的线性相关性良好,相关系数(R)均大于0.999;BPA、E1、EE2及E3的线性范围为3~300 ng/L,17α-E2及17β-E2的线性范围为5~300 ng/L;方法检出限为2.0~5.0 ng/L。
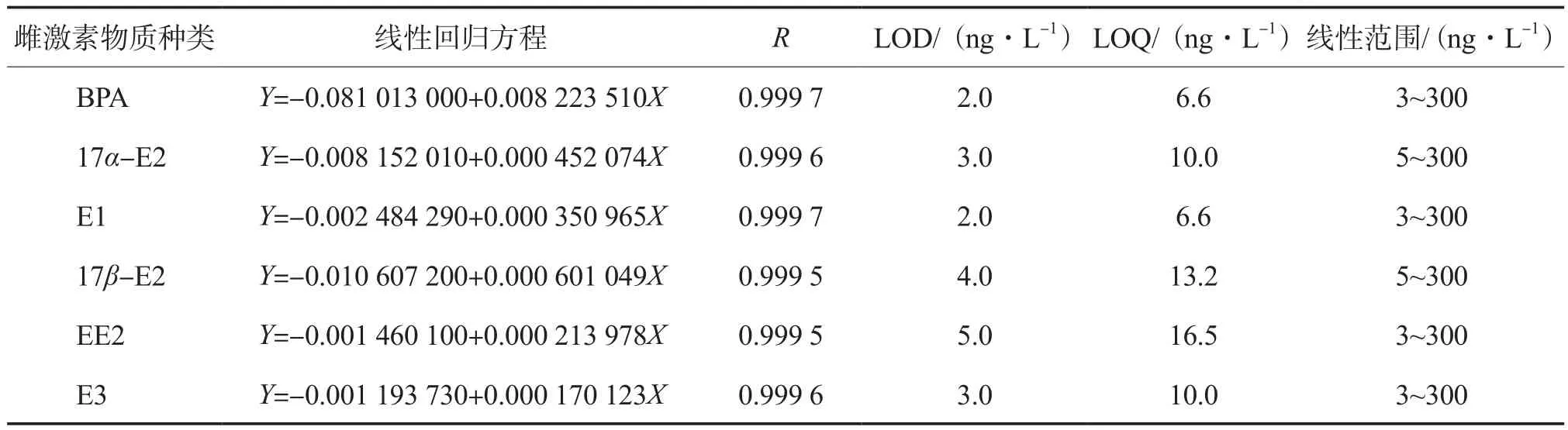
表3 6种雌激素物质的标准曲线及相关系数、检出限、定量限及线性范围
2.6 方法加标回收率及精密度
6种雌激素物质的回收率和相对标准偏差见表4。由表4可见,3种加标水平下的加标回收率范围为76.45%~96.24%,相对标准偏差RSD范围为3.2%~9.8%(n=3)。表明本方法的回收率较高,精密度良好。
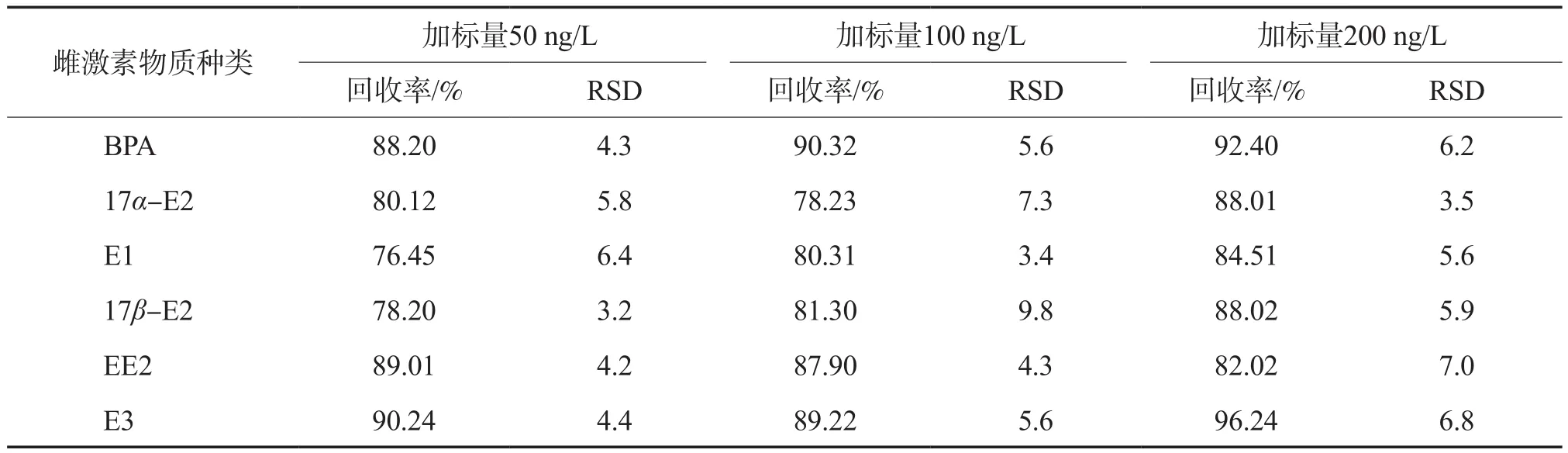
表4 6种雌激素物质的回收率和相对标准偏差(n=3)
2.7 与其他方法的比较
本方法与其他方法的对比见表5。由表5可见:本方法可以同时测定6种雌激素物质,分析物数量比其他3种方法更多;采用微波加热将衍生化时长缩短至4 min,相比其他方法,衍生化效率显著提升;同时还能保证方法检出限满足水样测定要求。综上,本方法灵敏快捷、简便高效,适用于大批量水样雌激素的分析测定。

表5 本方法与其他方法的对比
2.8 实际水样分析
采用建立的分析方法测定了云南大理洱海流域3处地表水样,测定结果见表6。由表6可见:除17β-E2外,其余5种雌激素物质均被检出,浓度范围为7.24~128.64 ng/L;其中17α-E2的流域浓度最低,为7.24~10.88 ng/L;3处水样中,水样#285的E1浓度显著高于其他两处水样,达到128.64 ng/L。
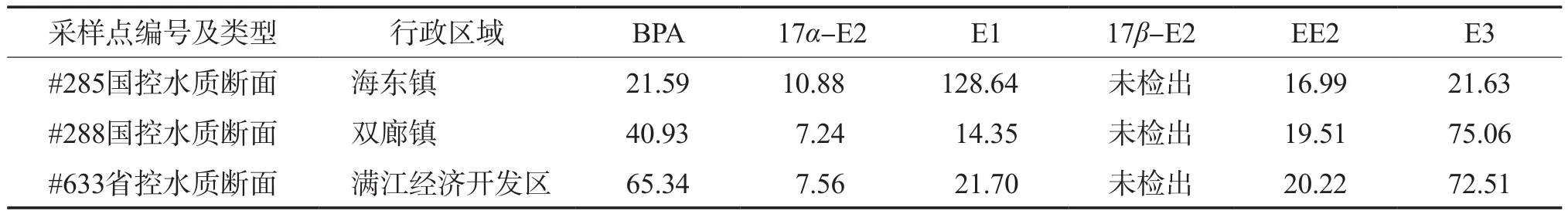
表6 实际水样的分析结果 ρ,ng/L
3 结论
a) 通过对气相色谱柱升温程序、前处理步骤中的固相萃取及衍生化条件进行优化,建立了可快速同时测定天然雌激素物质雌酮、17α-雌二醇、17β-雌二醇、雌三醇以及人工合成雌激素17α-乙炔基雌二醇、双酚A等6种雌激素物质的固相萃取—微波衍生化—GC-MS分析方法。
b)色谱柱最佳升温程序为:初始温度50 ℃,保持2 min;以20 ℃/min速率升至260 ℃,保持5 min;最后以10 ℃/min速率升至280 ℃,保持5 min。6种雌激素物质的全扫描色谱图的出峰效果良好,且分离度高。
c)最佳衍生化条件为:在试样中添加BSTFA+1%TMCS+吡啶作为衍生化试剂,315 W微波加热4 min。相比烘箱、水浴等传统衍生化加热方式,微波加热时间缩短至4 min,衍生化效率提升明显。
d)最佳固相萃取小柱类型为HC-C18小柱,回收率范围为82.93%~115.00%。本方法对BPA、E1、EE2及E3的线性范围为3~300 ng/L;17α-E2及17β-E2的线性范围为5~300 ng/L;方法检出限为2.0~5.0 ng/L,加标回收率为76.45%~96.24%。
