盐酸达泊西汀的合成及工艺优化
2020-04-20付丙月张宁张宗磊段崇刚
付丙月 张宁 张宗磊 段崇刚

摘 要 目的:优化盐酸达泊西汀的合成工艺。方法:采用手性合成的方法,以3-氯苯丙酮为原料,采用(1S,2R)-(-)-1-氨基-2-茚醇为催化剂、硼烷-N,N-二乙基苯胺为还原剂进行不对称还原,然后依次经过与α-萘酚醚化、磺酸酯化、二甲胺取代、HCl成盐反应得到最终产物。通过核磁共振和质谱技术对合成产物进行表征。对中间体Ⅰ、中间体Ⅱ、中间体Ⅲ和最终产物的合成反应进行工艺优化。结果:表征结果显示最终产物为盐酸达泊西汀,纯度为99.8%,收率为58.9%。与传统的拆分工艺比较,本工艺采用手性合成的方法不需要拆分,收率明显高于文献报道的拆分工艺收率(31.9%)。优化后的工艺减少了杂质的产生,提高了产品质量。结论:本合成工艺反应条件较温和、合成路线较短、收率较高。
关键词 盐酸达泊西汀;手性合成;不对称还原;工艺优化
ABSTRACT OBJECTIVE: To optimize the synthesis process of dapoxetine hydrochloride. METHODS: By chiral synthesis, asymmetric reduction was carried out by using 3-chlorophenylacetone as raw material, (1S, 2R) -(-)-1-amino-2-indanol as catalyst, and borane-N, N-diethylaniline (DEANB) as reducing agent. Then, it was reacted with α-naphthol etherification, sulfonation, dimethylamine substitution, and HCl salt formation reaction to obtain the final products. The products were characterized by NMR and MS. The synthesis reaction of intermediate Ⅰ, intermediate Ⅱ, intermediate Ⅲ and the final product were optimized. RESULTS: The final product was dapoxetine hydrochloride with purity of 99.8% and yield of 58.9%. Compared with traditional splitting technology, the chiral synthesis technology of this study did not need splitting, and the yield of the technology was significantly higher than that of splitting technology reported in literature (31.9%). The optimized technology reduced the generation of impurities and improved the product quality. CONCLUSIONS: The improved technology has milder reaction conditions, shorter synthesis route and higher yield.
KEYWORDS Dapoxetine hydrochloride; Chiral synthesis; Asymmetric reduction; Technology optimization
鹽酸达泊西汀(Dapoxetine hydrochloride)化学名为(S)-(+)-(N,N-二甲胺基)-3-(萘基-1-氧基)-1-苯基丙烷盐酸盐,是一种选择性5-羟色胺再摄取抑制剂(SSRIs),2009年在欧洲上市,之后陆续在多个国家和地区上市,2013年进入中国市场(商品名:必利劲),用于成年男子早泄的按需治疗,是世界上第一个被批准用于治疗早泄的口服药[1]。与传统的SSRIs相比,盐酸达泊西汀半衰期短、副作用小,具有更好的安全性和耐受性[2-3]。
盐酸达泊西汀的合成方法主要包括拆分法和手性合成法。本研究在已报道的合成方法基础上对盐酸达泊西汀的合成工艺进行了改进,使用3-氯苯丙酮为原料,采用(1S,2R)-(-)-1-氨基-2-茚醇为催化剂、硼烷-N,N-二乙基苯胺为还原剂进行不对称还原[4],然后经与α-萘酚醚化、磺酸酯化、二甲胺取代、HCl成盐反应制得最终产物,并通过核磁共振(NMR)和质谱(MS)技术对合成产物进行表征,以期缩短盐酸达泊西汀的合成路线,提高收率。
1 材料
1.1 仪器
1260型高效液相色谱(HPLC)仪和Q Exactive focus 型液质联用仪(美国Agilent公司);AVANCE Ⅲ型超导NMR波普仪(瑞士Bruker公司);WZZ-1型自动指示旋光仪(上海光学仪器修理厂)。
1.2 药品与试剂
3-氯苯丙酮、甲萘酚(上海嘉辰化工有限公司,化学纯);(1S,2R)-(-)-1-氨基-2-茚醇(山东威智医药工业有限公司,化学纯);硼烷-N,N-二乙基苯胺(阿拉丁试剂上海有限公司,分析纯);二甲胺2M四氢呋喃溶液(济南金贵林化工有限公司,医药级);4-二甲氨基吡啶、甲苯、乙酸乙酯、丙酮、二氯甲烷(国药集团化学试剂有限公司,分析纯);无水硫酸钠(天津市恒兴化学试剂制造有限公司,分析纯);N,N-二甲基甲酰胺(DMF)、正己烷(天津市康科德科技有限公司,分析纯);四氢呋喃、三乙胺(天津富宇精细化工有限公司,分析纯);水为自制纯化水。
2 合成与表征
2.1 合成路线
盐酸达泊西汀的合成路线详见图1。
2.2 R-(+)-3-氯苯丙醇(中间体Ⅰ)的合成
将(1S,2R)-(-)-1-氨基-2-茚醇(14.3 g,0.096 mol)、四氢呋喃(300 mL)加入到三口瓶中,降温至-5~0 ℃,搅拌1 h,缓慢滴加硼烷-N,N-二乙基苯胺(123.6 g,0.76 mol)溶液,滴毕继续保温1 h;然后滴加3-氯苯丙酮(162.0 g,0.96 mol)的四氢呋喃(800 mL)溶液,滴毕反应3 h;再缓慢滴加丙酮(200 mL),滴毕反应1 h。减压浓缩蒸出溶剂,加入二氯甲烷(800 mL),用10%的硫酸(400 mL×2)洗涤二氯甲烷,有机层经无水硫酸钠干燥,过滤,滤液减压蒸干,加入正己烷,室温搅拌,过滤,得到白色固体154.0 g,收率为95.0%。经测定,熔点(mp):56~57 ℃;纯度为93.8%(HPLC,面积归一化法)[5];比旋光度[α]23D=+25.3,c=1,CHCl3;光学纯度(ee):98.8%;1H-NMR(400 MHz,CDCl3),δ:2.01(s,1H,OH),2.20~2.37(m,2H,CH2),3.38~3.80(m,2H,CH2),4.69(dd,1H,J=4.8、8.4 Hz,CH),7.19~7.40(m,5H,ArH);MS,质荷比(m/z):171.6(M+)。
2.3 (R)-(-)-3-(1-萘氧基)-1-丙醇(中间体Ⅱ)的合成
将缚酸剂氢氧化钾(36.5 g,0.65 mol)、DMF(160 mL)加入到三口瓶中,降温至5 ℃左右,缓慢滴加含甲萘酚(93.7 g,0.65 mol)的DMF溶液(200 mL),搅拌1 h,滴加含中间体Ⅰ(100 g,0.59 mol)的DMF溶液(100 mL),升温至35~40 ℃继续反应20 h。反应完毕后,向反应液中加入800 mL水,然后用甲苯(500 mL×3)提取,合并甲苯层,依次用5%的氢氧化钠水溶液、纯化水、饱和氯化钠溶液洗涤后,再经无水硫酸钠干燥,减压蒸干,得到淡棕色油状物。向油状物中加入正己烷(600 mL),剧烈搅拌2 h析出固体,过滤,滤饼干燥后得到灰白色固体140.9 g,收率:86.4%。经测定,纯度:98.3%(HPLC,面积归一化法);1H-NMR(400 MHz,CDCl3),δ:1.86(s,1H,OH),2.25~2.30(m,2H,CH2),3.50~3.92(m,2H,CH2),4.56~4.62(m,1H,OCH),6.78(d,1H,J=7.5 Hz,ArH),7.19~7.76(m,10H,ArH),7.19~7.40(m,5H,ArH);MS,m/z:279.3(M+)。
2.4 (S)-(+)-N,N-二甲基-α-[2-(萘氧基)乙基]苯甲胺(中間体Ⅲ)的合成
将中间体Ⅱ(110 g,0.40 mol)、三乙胺(60.7 g,0.60 mol)、4-二甲氨基吡啶(4.9 g,0.04 mol)、四氢呋喃(1 000 mL)加入到三口瓶中,氮气保护下降温至0~5 ℃,滴加甲磺酰氯(91.98 g,0.48 mol),滴毕后,保持0~5 ℃反应1 h。向反应瓶中加入二甲胺2M四氢呋喃溶液(220 mL,0.44 mol),25 ℃下搅拌15 h。减压回收溶剂,残余物中加入水(1 000 mL)、二氯甲烷(1 000 mL)萃取,有机层用水(500 mL)洗涤,以无水硫酸钠干燥,过滤,滤液减压浓缩得到棕色油状物90.6 g,收率为74.2%。经测定,纯度:99.6%(HPLC,面积归一化法);MS,m/z:306.4(M+)。
2.5 盐酸达泊西汀的合成
向中间体Ⅲ中加入乙酸乙酯(400 mL),降温至0~5 ℃,向反应液中通入干燥HCl气体至pH为1.0,室温搅拌1 h,过滤,得到白色固体99.6 g,收率为98.0%。经测定,纯度:99.8%(HPLC,面积归一化法);mp:178~180 ℃;比旋光度[α]23D=+129~130,c=1,MeOH;1H-NMR(400 MHz,CDCl3),δ:2.23(s,6H,NMe2),2.25~2.30(m,1H,CH2),2.50~2.70(m,1H,CH2),3.50~3.60(m,1H,Me2NCH);3.82~4.15(m,1H,CH2),6.55~6.75(d,1H,J=7.5 Hz,ArH),7.28~7.60(m,7H,ArH),7.52~7.60(m,2H,ArH),7.78(d,1H,J=8.7 Hz,ArH),8.22(d,1H,J=8.4 Hz,ArH),12.9(s,1H,HCl);13C-NMR(300 MHz DMSO-d6)δ:36.5,38.9,39.0,61.2,67.9,103.6,120.2,121.9,125.0,125.5,125.7,125.8,126.3,127.6,128.3,128.4,128.6,128.7,134.5,139.1,155.7;MS,m/z:306.2(M+)。
3 工艺优化要点分析
3.1 中间体Ⅰ的合成工艺优化
中间体Ⅰ的合成是盐酸达泊西汀合成的关键步骤,已报道的文献方法[6-7]利用NaBH4-氨基酸衍生物进行不对称还原,所得产物需要柱层析分离纯化,且ee值不高。本研究采用硼烷-N,N-二乙基苯胺进行不对称还原,合成了ee值高达98.8%的中间体Ⅰ,然后重结晶精制,更加适合工业化生产。此外,本研究还考察了反应温度(0~5、-5~0、-10~-5 ℃)和反应时间(2.5、3、3.5 h)对中间体Ⅱ合成的影响,结果显示,随着反应温度的降低,中间体Ⅰ的收率变化不大,但-5~0 ℃时,ee值明显高于0~5 ℃时的ee值,但与-10~-5 ℃时比较ee值变化不大。随着反应时间的延长,中间体Ⅰ的收率逐渐增加,ee值先增加后降低,当反应时间为3 h时,收率较高,ee值最高。综合考虑,不对称还原反应的温度选择为-5~0 ℃,反应时间为3 h。反应温度对中间体Ⅰ收率和ee值的影响详见表1,反应时间对中间体Ⅰ收率和ee值的影响详见表2。
3.2 中间体Ⅱ的合成工艺优化
甲萘酚与中间体Ⅰ发生缩合反应,生成中间体Ⅱ。本研究分别考察了甲萘酚-中间体Ⅰ不同物料比(0.9、1.0、1.1、1.2,mol/mol)和不同缚酸剂(氢氧化钠、氰化钠、氢氧化钾)对中间体Ⅱ合成的影响。结果显示,随着物料比的增大,中间体Ⅱ的收率逐渐增加,HPLC法测得的纯度呈现先增加后降低的趋势。其中当甲萘酚-中间体Ⅰ物料比为1.1时,中间体Ⅱ的收率和纯度最高;当使用氢氧化钠为缚酸剂时,其收率和纯度均较低;当氰化鈉为缚酸剂时,其收率和纯度均有所提高;而使用氢氧化钾为缚酸剂时,其收率和纯度均最高。因此,中间体Ⅱ合成时选择甲萘酚-中间体Ⅰ的物料比为1.1,缚酸剂为氢氧化钾。物料比对中间体Ⅱ收率和纯度的影响详见表3,缚酸剂对中间体Ⅱ收率和纯度的影响详见表4。
3.3 中间体Ⅲ的合成工艺优化
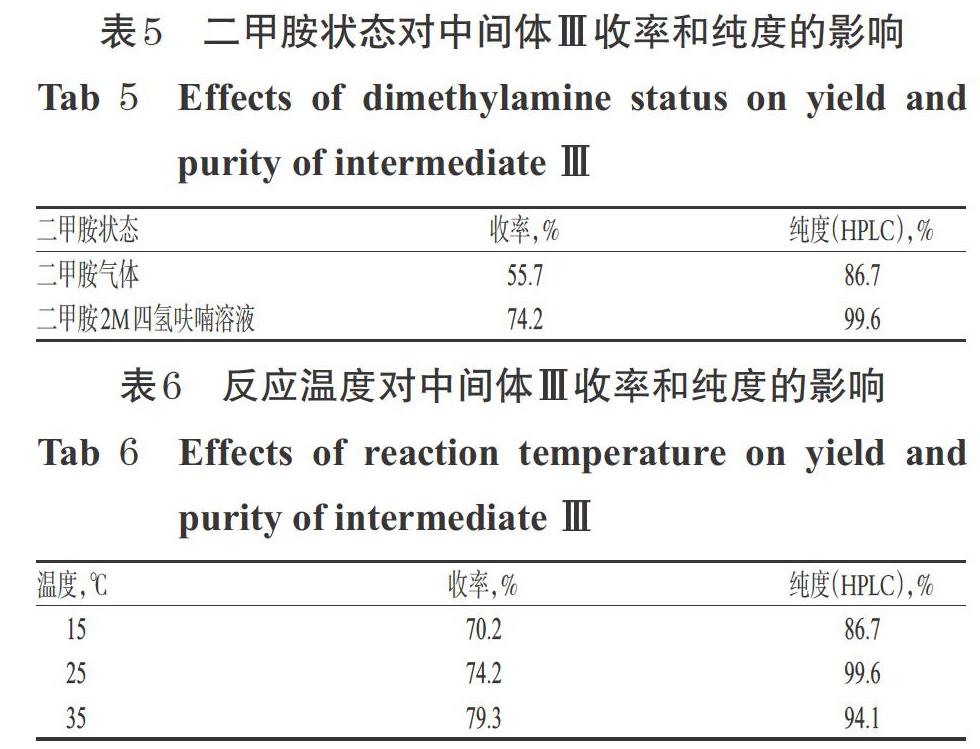
按“2.4”项下方法,中间体Ⅱ依次进行磺酸酯化、二甲胺SN2亲核取代,生成中间体Ⅲ。本研究考察了反应底物二甲胺的状态(气态和液态)和反应温度(15、25、35 ℃)对中间体Ⅲ合成的影响。结果显示,采用二甲胺气体(由33%二甲胺水溶液加热、干燥制备)作为反应底物时,中间体Ⅲ的收率和HPLC法测得的纯度均较低,且反应过程中很难定量控制,如果通气量少会使反应不完全,如果通气量过大则会造成浪费和环境污染;而采用二甲胺2M四氢呋喃溶液为反应底物时,则能准确控制二甲胺的投料量,避免浪费和污染,合成的中间体Ⅲ的收率和纯度也均高于前者;随着反应温度的升高,中间体Ⅲ的收率逐渐增加,纯度先增加后降低;当反应温度为25 ℃时,收率较高,纯度最高。综合考虑,选择二甲胺2M四氢呋喃溶液作为该工艺的反应底物,反应温度为25 ℃。二甲胺状态对中间体Ⅲ收率和纯度的影响详见表5,反应温度对中间体Ⅲ收率和纯度的影响详见表6。
3.4 盐酸达泊西汀的合成工艺优化
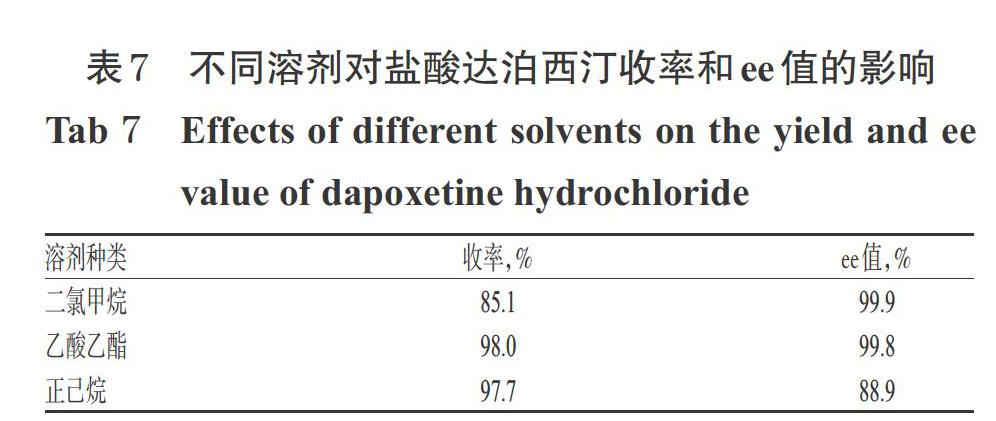
按“2.5”项下方法,中间体Ⅲ的溶液中通入HCl气体,成盐反应生成盐酸达泊西汀。本研究考察了中间体Ⅲ的不同溶剂(二氯甲烷、乙酸乙酯、正己烷)对盐酸达泊西汀合成的影响。结果显示,溶剂为二氯甲烷时,盐酸达泊西汀的ee值最高,但收率最低;溶剂为正己烷时,盐酸达泊西汀的收率较高,但ee值最低;而乙酸乙酯作为溶剂时,盐酸达泊西汀的收率最高,ee值也较高。综合考虑,选择乙酸乙酯作为成盐反应的溶剂。不同溶剂对盐酸达泊西汀收率和ee值的影响见表7。
4 讨论
盐酸达泊西汀的合成方法中,传统的拆分法[6-14]需多次拆分才能得到手性纯度较高的产品,操作繁琐,成本较高。2019年最新文献报道中,孙铭等[10]以3-氯-1-苯丙酮为起始原料,经过还原、取代、酯化、胺化、拆分、成盐反应得到盐酸达泊西汀的合成路线的收率为31.9%,产物纯度99.7%;王福生等[11]以苯丙醇和萘酚为原料,经6步反应合成盐酸达泊西汀,合成路线的总收率为17.2%,产物纯度为99.9%。这两条合成路线虽然所获产物纯度比较高,但是收率均较低,不适合工业化大生产。
盐酸达泊西汀的合成方法中手性合成法主要有3条路线:(1)以3-氯苯丙酮为原料[15-16],采用α-蒎烯类手性硼烷还原剂进行不对称还原,然后经过醚化、甲磺酰化、胺取代、成盐反应制得盐酸达泊西汀。该方法用到的α-蒎烯类手性硼烷易燃易爆,操作危险,并需要-24 ℃低温反应,反应条件苛刻,且不对称还原收率不高。(2)以N-Boc-(R)-苯甘氨酸为起始原料[17],经硼烷还原、甲磺酸酯化、氰化钠取代、水解、羧基还原、胺甲基化、缩合7步反应制得盐酸达泊西汀。该方法所用原料价格高不易得,反应步骤冗长,而且用到剧毒试剂氰化钠。(3)以肉桂酸酯为原料[18],经Sharpless不对称双羟基化、Barton-McCombie脱氧剂Mitsunobu反应、氨基甲基化和取代等多步反应制得盐酸达泊西汀。该方法步骤较长,操作繁琐,总收率仅为17%。
本研究以3-氯苯丙酮为起始原料,经硼烷-N,N-二乙基苯胺不对称还原[16]、甲萘酚缩合、磺酸酯化、二甲胺SN2亲核取代,通HCl气体得到目标产物。与上述文献报道的合成工艺比较,本工艺路线合成的盐酸达泊西汀的光学纯度极高(ee>98%),且总收率较高(58.9%)。同时采用二甲胺2M四氢呋喃代替二甲胺气体,可准确控制二甲胺的投料量,且操作简单,避免浪费。因此,本方法具有原料易得、操作简单、反应条件较温和、分离纯化操作简单、收率和产品纯度高等优点。
参考文献
[ 1 ] 张建忠,李红军.早泄治疗的新进展[J].中华男科学杂志,2018,24(10):933-936.
[ 2 ] 侯瑞鹏,李建,李昭夷,等.早泄标记物的应用研究[J].临床泌尿外科杂志,2020,35(1):10-13.
[ 3 ] 邓玉晓,孙晋瑞,段崇刚,等.联苯乙酸合成工艺的优化研究[J].中国药房,2018,29(20):2768-2772.
[ 4 ] 百灵,肖鸽,卓广澜. CBS催化硼烷氨配合物不对称还原法制备(S)-3-氯-1-苯基-1-丙醇[J].应用化学,2012,29(9):1087-1089.
[ 5 ] 冯光玲,冯爱国,丁文娟,等.一种达泊西汀中有关物质的高效液相色谱法测定方法,中国:CN103926335A[P]. 2014-07-16.
[ 6 ] 武平华,杜全海,陆涛.盐酸达泊西汀的合成[J].中国现代应用药学,2016,33(2):181-183.
[ 7 ] 白洁,曲辉,吴静,等. 3-(1-萘氧基)-1-苯基丙醇的合成工艺改进[J].当代化工研究,2018(2):157-158.
[ 8 ] 蒋强华,徐永祥,杨洋,等.达泊西汀的合成方法研究进展[J].中国药物化学杂志,2013,23(5):417-421.
[ 9 ] 王亚娜,陶莹莹,张恩.达泊西汀合成研究进展[J].广东化工,2015,42(6):88-89.
[10] 孙铭,白洁,张作鹏,等.盐酸达泊西汀的合成工艺改进[J].中国药物化学杂志,2019,29(2):122-125.
[11] 王福生,蘇艳华.盐酸达泊西汀的合成与表征[J].化工技术与开发,2019,48(11):44-46.
[12] OLIVER T,VICENTE GF,VICENTE G. Lipase-catalyzed resolution of chiral 1,3-amino alcohols:application in the asymmetric synthesis of (S)-dapoxetine[J]. Tetrahedron:Asymmetry,2006,17(5):860-866.
[13] 郭建敏,杨锐生,杜曦,等.达泊西汀关键中间体的合成[J].泸州医学院学报,2013,36(1):47-48.
[14] 薛大泉,张威,邓泽军,等.盐酸达泊西汀的合成[J].合成化学,2010,18(5):647-649.
[15] MAHALE RD,CHASKAR,SP,PATIL KE. Corey-itsuno reduction of ketones:a development of safe and inexpensive process for synthesis of some API intermediates[J]. Org Process Res Dev,2012,16(4):710-713.
[16] 宁岩石,临长江,路志进,等.达泊西汀的合成[J].化学试剂,2015,37(2):185-188.
[17] SHAFI AS,KUMAR VS. Enantioselective synthesis of (S)-dapoxetine[J]. Elsevier Tetrahedron:Asymmetry,2007,18(17):2099-2103.
[18] BROWN HC. Novel process of producing phenyl or substituted phenylakylamine pharmaceutical agents and novel chiral intermediates of high enantiomeric purity useful therein:US,4868344A[P]. 1989-09-19.
(收稿日期:2019-12-31 修回日期:2020-02-21)
(编辑:邹丽娟)
