西伯利亚白刺基因组信息初探
2020-04-16朱礼明黎梦娟张景波杨秀艳成铁龙
朱礼明,黎梦娟,张景波,杨秀艳,4,成铁龙*
(1. 南京林业大学,林木遗传与生物技术省部共建教育部重点实验室,江苏 南京 210037;2. 南京林业大学南方现代林业协同创新中心,江苏 南京 210037;3. 中国林业科学研究院沙漠林业实验中心,内蒙古 磴口 015200;4. 中国林业科学研究院国家林业和草原局盐碱地研究中心,北京 100091)
西伯利亚白刺(Nitraria sibirica Pall)系蒺藜科白刺属植物,为第三纪孑遗植物,分布于蒙古、中亚以及我国西北、华北、东北的沙地、盐碱地地区[1]。西伯利亚白刺具耐盐碱、抗风沙等特性,能在沙漠盐碱等恶劣环境下生存,是一种优良的沙地、盐碱地改良物种,其果实富含多种氨基酸、糖类、黄酮等物质[2-4],营养价值丰富,其地上部分也可作为牲畜饲料。因此,西伯利亚白刺兼有生态和经济价值,有较好的开发利用前景。
目前,关于西伯利亚白刺的研究主要集中在繁殖技术优化[5-6]、果实成分测定[7-8]及生理生化测定[9-11]等方面,有关西伯利亚白刺的分子生物学方面的研究较少[12],基因组学方面的研究也尚未见报道。宏观的研究只能从表层揭示西伯利亚白刺抗逆适应现象 ,并不能从内部机制、进化等层面解释西伯利亚白刺抗逆机理,而全基因组测序可以获取典型基因组特征并获得大量基因序列,对于剖析其生长、发育、抗逆等机理,发掘西伯利亚白刺的生态和经济价值有积极意义[13-14]。
全基因组调查通过了解待测生物基因组的基本特征,可以对全基因组测序组装难度、组装时间和成本等作出大致的评估并作出相应的测序策略调整,是基因组测序前必不可少的步骤之一。
流式细胞术是一种快速预测基因组大小的技术,它通过比较待测植物和标定植物细胞悬液荧光吸收峰相对比值,再根据标定植物的基因组大小来计算待测植物基因组大小[15]。而随着基因组测序技术的成熟及成本的下降,通过全基因组survey来探究待测植物的基因组基本特征不失为一种有效的方法,作为近年来发展较快的基因组预测技术,全基因组survey可以对生物的基因组基本特征测定评估[16-17],相比于流式细胞术等基因组大小预测方法,不仅可以精准预测基因组大小,还可以对基因组复杂程度、杂合率、重复序列比例等有相应的评估,更能切合生物的基因组特征,因而有更好的参考价值。
SSR分子标记以其高重复性、高多态性、共显性遗传、丰度高等优良特性成为了研究群体遗传学、遗传变异和标记辅助选择的有力工具,对于了解西伯利亚白刺的进化有积极的作用。
本研究基于流式细胞术和全基因组survey测序的方法对西伯利亚白刺基因组大小、复杂程度、杂合率等基因组特征有一个较为详细的评估,同时也对其测序方案的制定提出建议,为后续西伯利亚白刺基因组组学研究奠定了良好的基础。
1 材料和方法
1.1 材料
将取自内蒙古磴口的野生西伯利亚白刺种子置于4℃下沙藏30 d,置于萌发盒上进行萌发,再将发芽的种子定植于7 cm×7 cm的塑料花盆中(基质配方为河沙∶营养土=1∶1,并在其中掺入少量珍珠岩和蛭石),幼苗生长2个月后取嫩叶备用。流式标定植物为 Jaroslav Dolezˇel博士惠赠的番茄‘Stupicke´polnı´ rane´’ 32 品种。
1.2 方法
1.2.1 流式细胞分析 使用BD公司influx型号流式细胞仪对西伯利亚白刺基因组大小进行分析,选用mG解离液对植物叶片进行解离,使用碘化丙啶(PI)溶液为荧光染料,采用本番茄作为内标,使用Influx自带分析软件FACSTM分析基因组大小。
操作步骤:于塑料皿上滴加1.5 mL mG解离液,分别取0.5 g西伯利亚白刺、番茄新鲜叶片用刀片迅速切碎后过400目滤网,将收集的滤液1 500 rpm,离心6 min,吸除上清液后重新加入500 μL预冷的mG解离液,加入PI染色液,最后加入10 μg·mL-1的 Rnase,避光 4℃ 孵育 5 min 后低速上机检测。
C值计算公式:C待测样本=C标定×(G0/G1待测样本/G0/G1标定)
式中:G0/G1为流式荧光吸收强度。
mG解离液配方:
45 mmol·L-1MgCl2,20 mmol·L-1MOPS, 30 mmol·L-1Na3C6H5O7·2H2O, 1%( w/v) PVP-40,0.2%(v/v)TritonX-100,10 mmol·L-1Na2EDTA,20 μL·mL-1β-巯基乙醇,调节 pH 至 7.0,-20℃ 下保存。PI为碘化丙啶,使用时至终浓度为50 μg·μL-1,4℃保存。
1.2.2 DNA的提取以及质量检测 采用CTAB法对西伯利亚白刺的新鲜叶片进行DNA提取,得到的DNA样品用紫外分光光度计检测其浓度、OD260/OD280,再经1%琼脂糖凝胶电泳检测其完整性(电泳条件为:电压180 V,电泳时间:30 min)。
1.2.3 文库制备及测序方法 检测合格的DNA样品通过Covaris超声波破碎仪打断成片段,并进行末端修复,加poly-A尾,加测序接头,纯化,PCR扩增等步骤后,构建出350 bp双端PE150待测序文库。文库通过Illumina Hiseq平台进行双端PE测序。
1.2.4 K-mer分析 采用K-mer分析策略,若每条序列的长度为L,K-mer长度为K,可以得到LK+1个K-mer,再通过这些数据来对基因组大小进行预估,通过Lander-waterman算法对西伯利亚白刺基因组大小进行估计,满足公式:

式中:Nbase和NK-mer为序列的碱基总数和K-mer数,Cbase和CK-mer为覆盖碱基的期望深度和K-mer期望覆盖深度。
对预估的基因组大小进行修正,将K-mer深度为1的情况认为是错误情况,计算错误率,并用于修正基因组大小,修正公式为

式中:Grevised为修正后的基因组大小,E为测序错误率。
通过K-mer数学分析模型,基因组杂合率公式为:
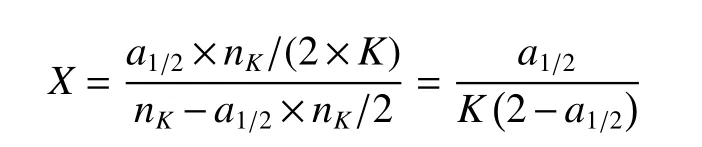
式中:a1/2为杂合K-mer种类数的百分比,nK为所有K-mer的种类数。
另外,计算标准泊松分布和实际数据曲线峰值后的面积差值,可得到重复序列百分比,在这里我们计算纯合峰深度1.8倍后面的K-mer个数所占的比例来估计重复序列比例。
1.2.5 基因组组装 由于西伯利亚白刺基因组重复序列较多,我们选择K-mer=41将打断的DNA序列拼接组装到Scaffold,通过reads之间的overlap关系构建de Bruiji图并对其简化,在重复区域边界位置进行剪切,得到contig序列,再根据大片段数据的Pair-end关系,构建Scaffold序列,最后用reads对Scaffold的gap区域进行填补,完成组装过程,具体配置参数为
pregraph : -K 41 -R -d 1
-K kmer: K value in kmer
-R (optional): unsolve repeats by reads (default no)
-d KmerFreqCutoff(optional): delete kmers with frequency no larger than (default 0)
contig : -D 1 -M 1 -R
-D EdgeCovCutoff(optional): delete edges with coverage no largert than (default 1)
-M mergeLevel (default 1,min 0, max 3): the strength of merging similar sequences during contiging
-R solve_repeats (optional): solve repeats by read paths(default: no)
map : -K 41
-K kmer (default: the same as in pregraph): k value in kmer
scaff : -F 1 -L 43
-F (optional) fill gaps in scaffold. (default 0;1:normally; -1:only fill nonrepeat gap; 2:radically)
-L minLen : shortest contig (minus K value) for scaffolding
再根据组装结果统计其contig分布情况,统计测序长度大于500 bp的测序深度和GC含量并做GC含量分布图。
1.2.6 SSR分布特征分析 运行MISA脚本(pgrc.ipk-gatersleben.de/misa)对过滤后数据SSR位点鉴定并统计其类型、数量。筛选标准为单核苷酸SSR位点≥16次,双核苷酸SSR位点≥6次,三四核苷酸SSR位点≥5次。
2 结果与分析
2.1 流式细胞基因组大小分析
将西伯利亚白刺和番茄的叶片混合解离液放入流式细胞运行并在480 nm波长下检测其荧光吸收强度(图1),其中,P0为西伯利亚白刺的吸收峰,P1为番茄的吸收峰,番茄参考2C值为1.96 pg,实验重复3次。将平均值代入C值计算公式得出:2C西伯利亚白刺=2C番茄×(G0/G1西伯利亚白刺)/(G0/G1番茄)=1.96 pg×0.534,得西伯利亚白刺C值大小为523.4 Mbp。
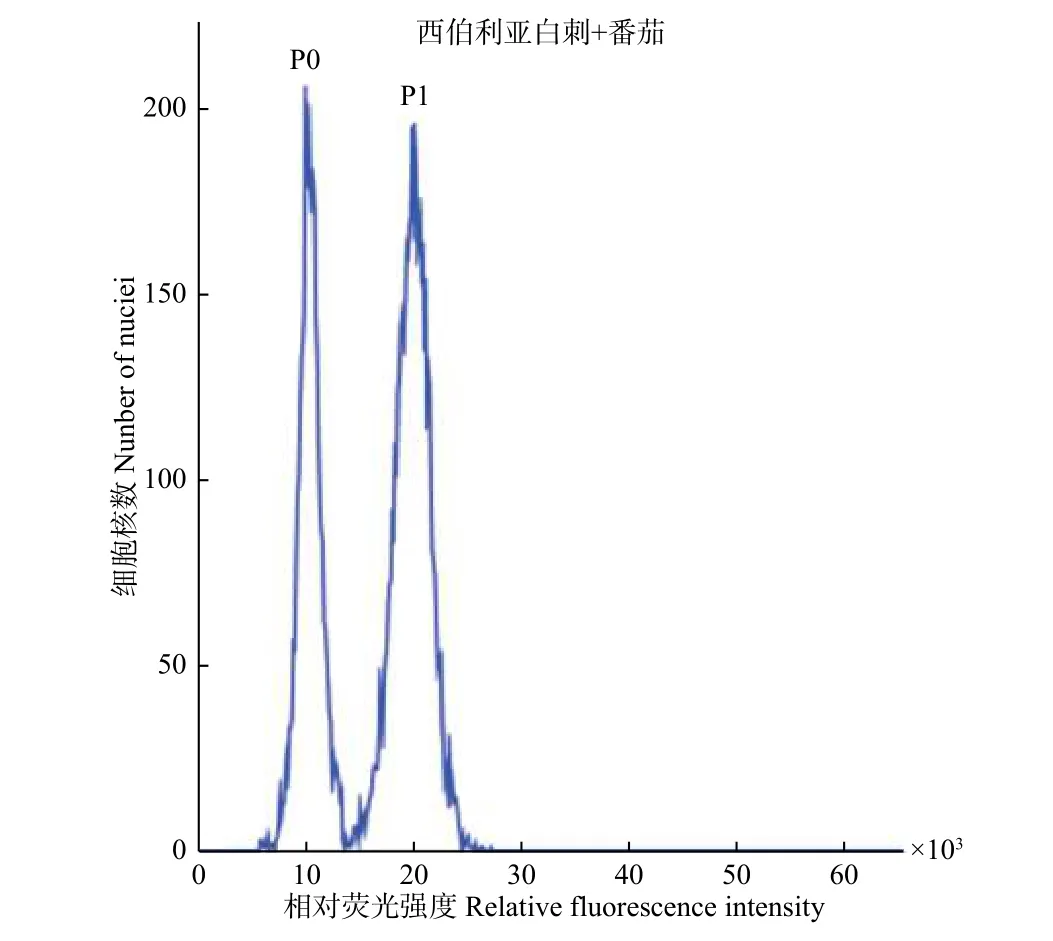
图 1 流式细胞测定结果Fig. 1 Flow cytometry results
2.2 DNA的提取以及质量检测
取1 μL DNA样品于分光光度计的检测,结果显示 OD260/OD280为 1.89,浓度为 206.9 ng·μL-1。再利用1%琼脂糖凝胶电泳检测其条带完整性,图2 表明:电泳条带单一,无明显杂带。综合二者推测,此DNA完整度较高,可用于下游实验。
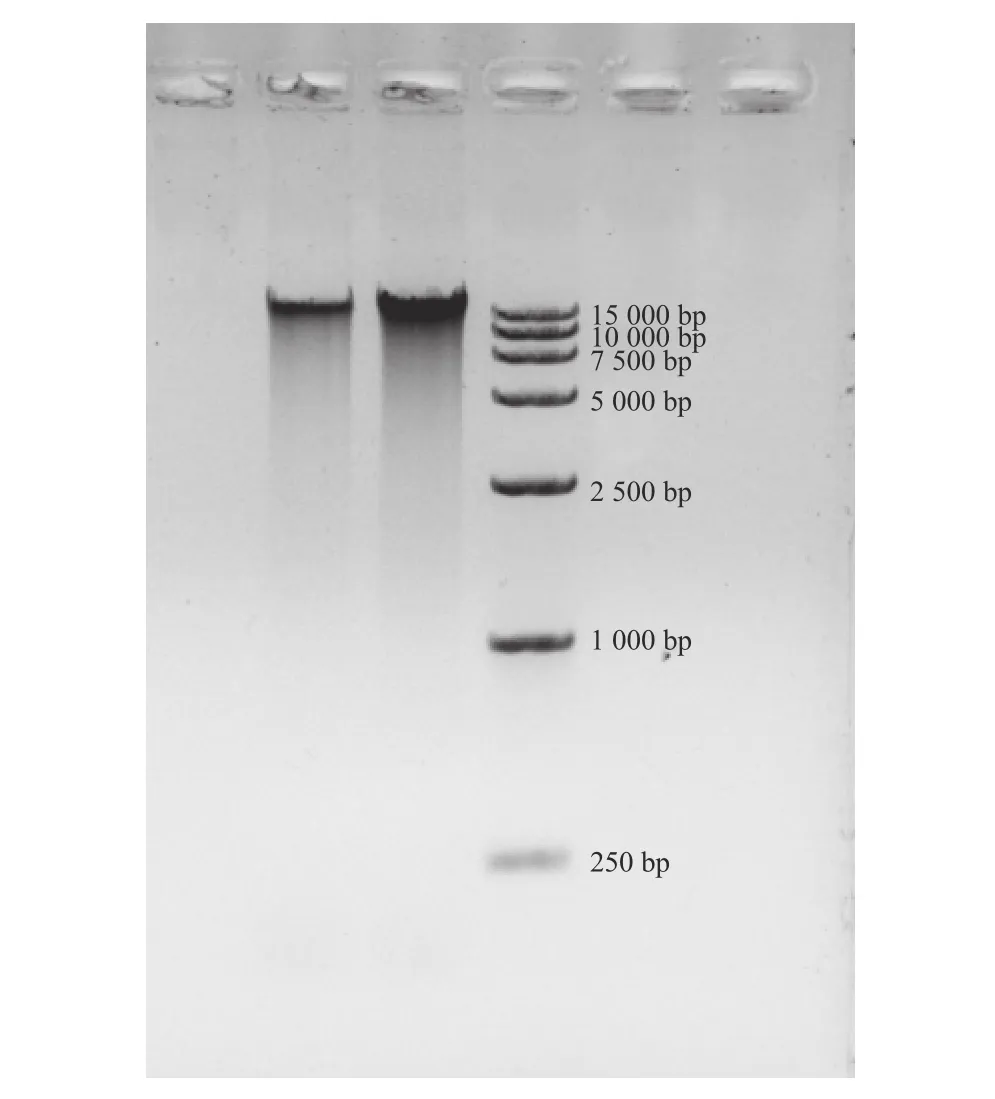
图 2 DNA琼脂糖凝胶电泳图Fig. 2 DNA agarose gel electrophoresis
2.3 测序数据产出及质控
2.3.1 测序数据统计 过滤掉无效或低质量的reads数据,再经图像识别、去污染等步骤,得出最终的测序结果(表1):其中,测序的总reads数为212 852 294个,测序的总数据大小为63 855.69 Mbp,按照536.16 Mbp的预估基因组大小得出本次测序深度为119.09×,测序的错误率为0.04%,Q20的含量为95.59%,Q30的含量为89.33%,GC含量为36.78%。

表 1 测序结果统计Table 1 Sequencing results statistics
2.3.2 测序质量检测 测序数据的质量主要分布在Q30(≥80%)以上,这样才能保证后续分析的正常进行,如图3所示,实验Q30含量为89.33%满足后续分析要求。
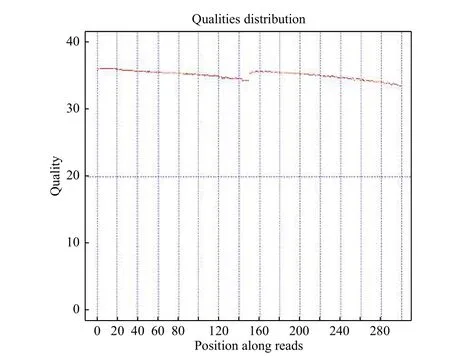
图 3 数据质量分布Fig. 3 Data quality distribution
此外,测序错误率也影响测序结果的准确性,对于下游分析至关重要,本实验2个reads的测序错误率均低于1%(图4),表明本次测序错误率控制良好。为进一步保证测序结果的可信性,还需对本次测序的碱基含量分布进行分析。GC含量分布检查用于检测有无AT、GC分离现象,理论上G和C含量以及A和T含量在每个测序循环上应分别相等,且整个测序过程中稳定不变,呈水平线。由于DNA模板扩增偏差等原因使测序前几个碱基测序质量值较低,发生小幅度波动,属于正常情况。本实验中(图5)测序的G和C的含量和A和T的含量接近也保证了测序的可信度。
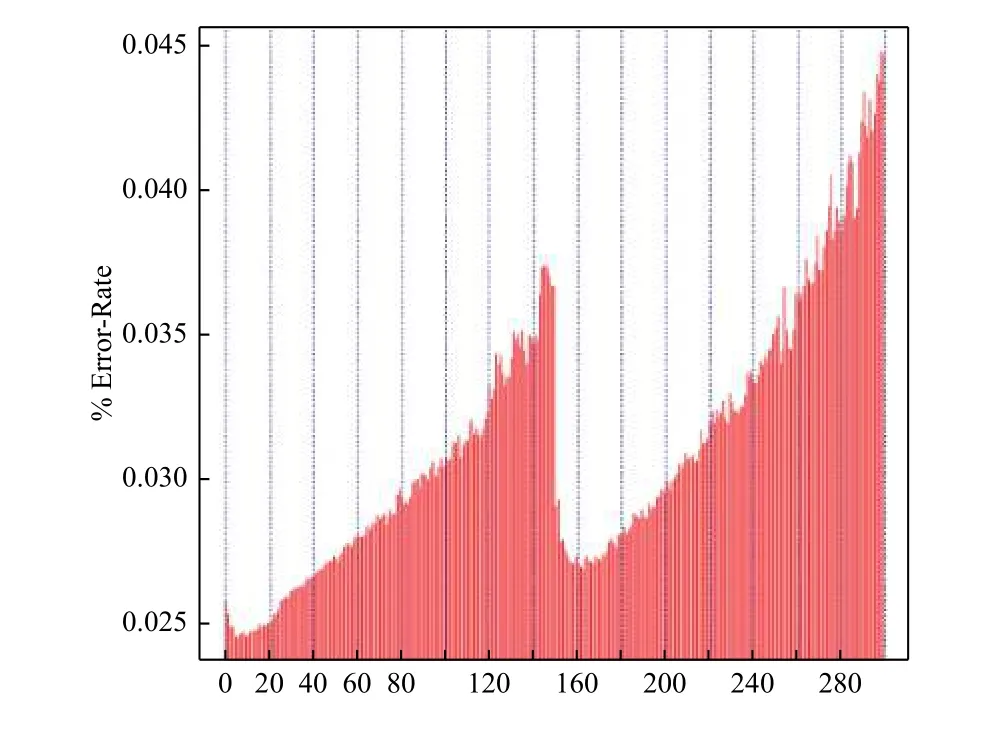
图 4 测序错误率分布Fig. 4 Sequencing error rate distribution

图 5 GC含量分布图Fig. 5 GC content distribution map
2.4 K-mer分析
利用K-mer分析法对西伯利亚白刺基因组大小进行估计,根据测序结果(表2、图6)发现:当K-mer深度为89×时存在明显的主峰,由K-mer相关公式计算得到的基因组大小为536.16 Mbp,并通过后续基因修正得修正后基因组大小为526.30 Mbp; 而在主峰前横坐标二分之一处出现次峰。一般当目标序列存在杂合现象时,存在杂合位点的K-mer被分成2份,频率变成原频率的1/2,因此,此峰为杂合峰,并统计得出西伯利亚白刺基因组杂合率为0.90%,杂合率较高,属于复杂基因组。此外,在约为主峰2倍depth的地方存在次峰,并有明显的拖带现象,该片段出现的期望值是大部分的2倍,这些片段为重复片段,由相关统计结果得重复序列数占总序列数的55.39%。

表 2 K-mer=17分析所得各项数据Table 2 K-mer=17 analysis of the data

图 6 K-mer=17 Depth和K-mer种类数频率分布图Fig. 6 K-mer=17 Depth and K-mer species frequency distribution
2.5 基因组组装
2.5.1 数据组装结果 运用Soapdenovo软件拼接上述测序数据,并对数据进行纠错,构建contig、scaffold等优化过程,得到初步的基因组组装信息(表3):针对组装好的长度大于等于100 bp的scaffold内部contig进行统计,得N50长度为1 076 bp,N90为 147 bp,组装得到最长的序列长度为45 660 bp,组装的contig总数量为917 423个,总长度为424 458 883 bp。进一步将所有文库测序得到的reads比对回初步得到的contigs,利用reads之间的连接关系和插入片段大小信息,过滤掉长度<100 bp的 contig序列,最终将 contigs组装成scaffolds,结果显示:N50的长度的1 889 bp,N90为189 bp,最长序列长度为89 063 bp,组装总量为717 232个,总长度为443 258 576 bp。

表 3 基因组组装结果统计Table 3 Genomic assembly results statistics
2.5.2 GC含量分布分析 GC含量是反映植物基因组成的重要指标之一,GC含量深度分析图用于检测测序是否存在GC分布偏向,样品是否存在细菌的污染等。由图7可得:西伯利亚白刺基因组测序没有明显的GC偏向。图中有2处GC聚集处,为了确认低测序深度区域是否为细菌污染造成,将低测序深度序列比对到NCBI核苷酸数据库,并没有细菌序列被比对上,说明样品没有被细菌污染,推测这是由于西伯利亚白刺基因组高杂合度所造成的。由于在组装过程中同源染色体上杂合部位只能被识别出一半,导致此部位的GC含量分布在低测序深度区域。

图 7 GC含量与测序深度关联分析统计图Fig. 7 GC content and sequencing depth correlation analysis
2.6 SSR位点分析
由MISA脚本分析西伯利亚白刺基因组数据并统计(表4),共搜寻到521 125个SSR位点,其中,单核苷酸位点出现比例最高,达342 883个,占总SSR位点的65.80%;二核苷酸位点146 312个,占比28.06%;三核苷酸位点26 133个,占5.02%;四个及以上核苷酸位点8 678个,占1.67%。所以,单核苷酸重复是西伯利亚白刺主要的SSR重复位点,同时单核苷酸重复中A/T占比最多,达到了63.94%。

表 4 西伯利亚白刺SSR位点统计Table 4 SSR locus statistics of N. sibirica
3 讨论
基因组大小是指生物单倍体染色体中DNA的含量,也称为C值[18]。目前为止已有数千种动植物的C值被检测并收录入相应的动植物C值库[19-20]。DNA的C值是生物体重要的基因特征,是种群分类的证据之一,也是开展各项基因工作的基础。了解基因组大小对于推测物种的演化趋势、进化地位、种属间进化关系、生物进化分类等具有深远的意义。
基因组大小预测常使用流式细胞术[21]、Feulgen图像分析法[22]、全基因组survey调查[23]等方法。流式细胞术通过比较待测植物和内标植物细胞悬液荧光吸收峰比值,根据公式由内标植物的基因组来计算待测植物基因组的大小,是一种快速、便捷的基因组预估的方法,在测定动植物体的基因组大小方面均有较广的应用。
全基因组survey测序是基于小片段文库的低深度从头测序,通过对原始数据进行图像识别,去污染、去接头等步骤,再进行K-mer分析,Soapdenovo软件组装继而完成整个分析过程,可对基因组的大小、GC含量、杂合率以及重复序列的含量等重要的基因组特征信息进行分析,相比于流式细胞仪、Feulgen图像分析法等基因组大小预测方法更能切合所测生物体基因组特征,是一种更精确的分析未知基因组特征的途径[24-26]。
西伯利亚白刺基因组GC含量为36.78%,没有明显的过高或过低的情况[27],对NGS测序准确性影响较小;而其杂合率为0.9%,基因组重复序列比例达55.39%,属于高杂合基因组。推测可能是由于西伯利亚白刺在地理分布上较广,生态条件悬殊、植物形态变化也较大有关[28]。
一般来说,基因组杂合度越大,重复片段越多,该物种的组装难度就越大。西伯利亚白刺属于高杂合基因组植物,而同为高杂合基因组的胡杨利用全基因组鸟枪法结合Fosmid拼装策略获得了精度较高的基因组图谱[29]具有一定参考意义,如果使用二代测序Platanus组装软件[30]可能更适合于西伯利亚白刺基因组的拼装。随着近年来测序成本的下降和3代测序技术的普及,二代llumina搭配三代Pacbio辅以Hi-C技术的方案将会是西伯利亚白刺全基因组测序更好的选择,更有利于获得高质量的全基因组图谱。
4 结论
本实验测得西伯利亚白刺基因组大小为536.16 Mbp,修正后为526.30 Mbp,杂合率为0.90%,重复序列比例为55.39%;西伯利亚白刺Contig N50为1 076 bp,总长为424 458 553 bp,Scaffold N50为1 889 bp,总长为443 258 576 bp。西伯利亚白刺有521 125个SSR位点,其中单核苷酸位点有342 883个,二核苷酸位点有146 312个,三核苷酸位点有26 133个,四个及以上为8 678个,单核苷酸为其主要的SSR特征。
