微波消解-原子荧光光谱法测定硫磺中微量砷的不确定度评定
2020-04-16涂振权汪玉洁欧阳清华鲁春
涂振权 汪玉洁 欧阳清华 鲁春
1.中国石油西南油气田公司天然气研究院 2.中国石油天然气质量控制和能量计量重点实验室
砷(As)含量是硫磺产品检测的重要项目,也是制约硫磺质量等级和用途的主要指标之一。目前,国内硫磺中As含量的检测主要依据的是GB/T 2449.1-2014《工业硫磺 第1部分:固体产品》规定的二乙基二硫代氨基甲酸银分光光度法[1]。该方法采用湿法消解,涉及溴和四氯化碳等多种化学试剂的使用,消解步骤繁琐,在操作时可能对人员健康和环境造成不利影响。
微波消解作为一种新兴的消解技术,是一种较安全、有效的样品前处理方法。它结合高压消解和微波快速加热两方面的性能,能消解许多湿法难以消解的样品,具有简单快速、试剂用量低、环境污染少、空白值低的优点,适合各类分析试样的前处理,尤其适合微量和痕量成分分析。同时,原子荧光光谱法具有谱线简单、灵敏度高、精确度高、操作简单等特点,已被广泛应用于工农业、生化制药、环境地质、食品安全等领域[2-8]。因此,采用微波消解-原子荧光光谱法测定硫磺中痕量As含量是一种技术发展趋势。目前,虽然对此测量方法已有文献报道[9-10],但均未对测量结果的不确定度进行评定。本方法根据JJF 1059.1-2012《测量不确定度评定与表示》和JJF 1135-2005《化学分析测量不确定度评定》对微波消解-原子荧光光谱法测定硫磺中微量As含量方法的测量不确定进行了评定[11-12],全面分析不确定度主要来源,为准确评价和使用检测结果提供依据。
1 实验
1.1 试剂和仪器
硝酸为优级纯,盐酸为原子荧光专用液,硫脲、抗坏血酸、硼氢化钾和氢氧化钠均为分析纯,实验用水为超纯水(≥18.2 MΩ·cm)。
As标准储备溶液:国家二级标准物质GBW(E) 080117, As单元素标准溶液(标准值100 μg/mL),相对扩展不确定度为0.8%(k=2),基体1%(体积分数)HNO3。
原子荧光光度计AFS-3100(北京海光仪器有限公司);As空心阴极灯;微波消解仪Multiwave 3000(安东帕公司);电子天平XS204(梅特勒公司)。
1.2 实验方法
1.2.1 样品消解和测定
称取约0.2 g硫磺,加3 mL硝酸,微波消解和微波赶酸,转移到50 mL容量瓶,再加入2.5 mL盐酸和10 mL 5%(质量分数)硫脲+5%(质量分数)抗坏血酸混合液,用超纯水定容至50 mL,摇匀后静置30 min以上,用AFS测定。同时做样品空白。测定流程见图1。

1.2.2 仪器工作条件
光电倍增管负高压280 V;As空心阴极灯电流50 mA;原子化器温度200 °C,原子化器高度8 mm;载气(Ar)流量400 mL/min;屏蔽气(Ar)流量900 mL/min;采用峰面积读数方式;读数时间10.0 s;延迟时间1 s;载液为5%(体积分数)盐酸;选用2.0%(质量分数)硼氢化钾和0.5%(质量分数)氢氧化钠的混合液作为还原剂。
1.2.3 标准曲线绘制
先将As标准储备溶液100 μg/mL稀释到1.00 μg/mL,再将1.00 μg/mL的As标准溶液配成As系列标准溶液(见表1)。

表1 As标准系列溶液V(1.00 μg/mLAs溶液)/mLV(盐酸)/mLV(5%硫脲+5%抗坏血酸混合液)/mLV最终/mLρ(As)/(ng·mL-1)0.002.510500.000.102.510502.000.202.510504.000.402.510508.000.502.5105010.00
2 结果与讨论
2.1 测量原理及数学模型
样品经微波消解和赶酸处理定容后,被转移到原子荧光光谱仪,在波长193.7 nm处进行荧光强度测量。基于荧光强度与As含量成正比。因此,通过与标准溶液荧光强度的比较得到定量。
硫磺中As质量分数按式(1)计算:
(1)
式中:w为硫磺中As质量分数,ng/g;C为测定液中扣除试剂空白后As的质量浓度,ng/mL;V为样品消化赶酸后的定容量,mL;m为样品质量,g。
2.2 不确定度来源分析
根据测量过程,本方法不确定度由待测样品的称量过程、消解过程、赶酸过程、定容过程和样品测定过程,以及标准溶液的纯度、稀释过程和AFS校准过程带来的,见图2和表2。


表2 不确定度来源分析不确定度来源标准不确定度相对标准不确定度硫磺样品的称量、消解和赶酸、定容样品均匀性称量过程(天平)u(m)urel(m)消解和赶酸:回收率u(Rec)urel(Rec)定容过程:体积u(V)urel(V)标准溶液纯度u(ρ)urel(ρ)稀释u(f)urel(f)AFS校准拟合曲线u(C)urel(C)结果的重复性测定算术平均值u(w测)urel(w测)
2.3 标准不确定度评定
2.3.1 样品制备过程引入的不确定度
2.3.1.1 取样(样品的均匀性)
本方法依据GB/T 6678-2003《化工产品采样总则》规定[13],在随机选定的每个采样单元中采样并充分混合均匀,然后依据GB/T 2449.1-2014《工业硫磺 第1部分:固体产品》规定方法制备样品[1],可认为样品是均匀的,代表性充分,由此引起的不确定度可忽略不计。
2.3.1.2 称量过程
称量所用的天平感量为0.1 mg,最大允差为0.000 2 g。按均匀分布,则由此带来的不确定度分量为:
天平称量两次(空瓶和空瓶+样品),所以由天平引起的标准不确定度为:
=0.000 163 g
称量0.2 g硫磺样品,由天平称量引入的相对不确定度:
2.3.1.3 消解回收率
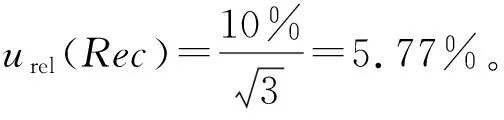
2.3.1.4 定容过程体积的影响
定容过程体积的影响包括容量瓶容量允差和温度引入的不确定度。
(1) 容量引起的不确定度:
所用的容量瓶为A级,标称容量50.00 mL,最大允差为0.05 mL,按均匀分布,则由此带来的不确定度分量为:
(2) 温度引起的不确定度:
实验室的温度在±5 °C之间变动,水的膨胀系数约为2.1×10-4,按均匀分布,则由此带来的不确定度分量为:
由定容过程引入的合成不确定度:
=0.041 9 mL
由定容过程引入的相对不确定度:
2.3.2 标准溶液不确定度
2.3.2.1 标准储备液的不确定度
As标准储备溶液采用中国计量科学研究院制备的国家二级标准物质GBW(E) 080117,As单元素标准溶液(标准值100 μg/mL),相对扩展不确定度为0.8%(k=2),基体1%(体积分数)HNO3。则As标准储备液的标准不确定度为:
2.3.2.2 稀释过程引入的不确定度
100 μg/mL As标准储备溶液经过两级稀释制成1.00 μg/mL As标准使用液,第一级稀释是将20 mL As标准储备液全部转移至200 mL容量瓶中,用水稀释并定容,制成10.00 μg/mL As标准溶液,第二级稀释是用20 mL移液管吸取20.00 mL 10.00 μg/mL As标准溶液,置于200 mL容量瓶中,用水稀释并定容至刻度,制成1.00 μg/mL As标准使用液。稀释过程引入的不确定度包括容量瓶体积带来的不确定度和移液管体积带来的不确定度。
(1) 容量瓶体积带来的不确定度包括容量瓶容量允差和温度引入的不确定度。
①容量引起的不确定度:
所用的容量瓶为A级,标称容量200.00 mL,最大允差MPE=0.15 mL,按均匀分布,则由此带来的不确定度分量:
②温度引起的不确定度:
实验室的温度在±5 °C之间变动,水的膨胀系数约为2.1×10-4,按均匀分布,则由此带来的不确定度分量为:
由容量瓶引入的合成不确定度为:
=0.149 mL
则由容量瓶引入的相对不确定度为:
(2) 移液管带来的不确定度包括移液管容量允差和温度引入的不确定度。
①容量引起的不确定度:
所用的移液为A级,标称容量20.00 mL,最大允差为0.030 mL,按均匀分布,则由此带来的不确定度分量:
②温度引起的不确定度:
实验室的温度在±5 °C之间变动,水的膨胀系数约为2.1 × 10-4,按均匀分布,则由此带来的不确定度分量:
由移液管引入的合成不确定度为:

=0.021 1 mL
由移液管引入的相对不确定度为:

二级稀释时稀释因子带来的不确定度为:
=0.001 30=0.130%
2.3.2.3 标准物质在一级稀释带来的不确定度
100 μg/mL As标准储备溶液标准物质经稀释制成10.00 μg/mL砷标准溶液,其不确定度由标准储备液的标准不确定和容量瓶体积引入的不确定度合成,即:
=0.00407=0.407%
2.3.2.4 标准物质在稀释过程中带来的不确定度
100 μg/mL As标准储备溶液经过两级稀释制成1.00 μg/mL As标准使用液,其不确定度由标准物质在一级稀释带来的不确定度和二级稀释时稀释因子带来的不确定度合成,即:
=0.427%
2.3.3 最小二乘法拟合标准曲线校准带来的不确定度
2.3.3.1 拟合曲线
采用6个含量水平的As标准溶液,用AFS测定,得到相应的荧光强度A,用最小二乘法拟合,得到直线方程和相关系数(见表3)。
设在分析条件下测量系列标准溶液的荧光强度,用最小二乘法回归得到工作标准曲线方程(一次一元线性方程),见式(2)。
A=bC+a
(2)
式中:A为荧光强度;C为溶液中As质量浓度,ng/mL;b为斜率;a为截距。

表3 As标准溶液含量与荧光强度关系表As标准溶液质量浓度C/(ng·mL-1)荧光强度A0.000.8012.00562.2294.001 163.9168.002 287.47610.002 875.744 注:标准曲线A=287.28C-0.89,相关系数r =1。

2.3.3.2 由标准曲线拟合带来的不确定度
标准曲线拟合带来的不确定度由式(3)计算:
(3)

残差标准偏差S由式(4)计算:
(4)
式中:A0i为各标准溶液荧光强度(a+b·C0i)为根据回归曲线计算的荧光强度。
则S=12.81。
=4.80 ng/mL
回归曲线质量浓度差的平方和:
标准曲线拟合带来的不确定度:
=0.032 9 ng/mL
计算数据及结果见表4。

表4 As标准工作曲线变动性的不确定度计算表序号As标准溶液质量浓度/(ng·mL-1)荧光强度回归曲线计算吸光度工作曲线吸光度差的平方10.000.801-0.892.8622.00562.229573.67130.9034.001 163.9161 148.23246.0548.002 287.4762 297.3597.50510.002 875.7442 871.9114.70求和24.00492.006被测样品的测量次数P67样品中As含量C1.288工作曲线校准点测量次数n59工作曲线各校准点质量的平均值4.810质量差平方和68.811S12.8112截距a-0.8913斜率b287.2814标准不确定度u(C)0.032 915相对标准不确定度urel(C)2.57%
2.3.4 结果的重复性带来的不确定度
在重复性条件下,连续测定6次,分别测得As的质量分数为:299 ng/g、301 ng/g、309 ng/g、309 ng/g、309 ng/g和318 ng/g,则测量的不确定度为:

测量结果的相对不确定度为:

2.4 合成相对标准不确定度
汇总第2.3节的评定数据,本方法不确定度分量见表5。
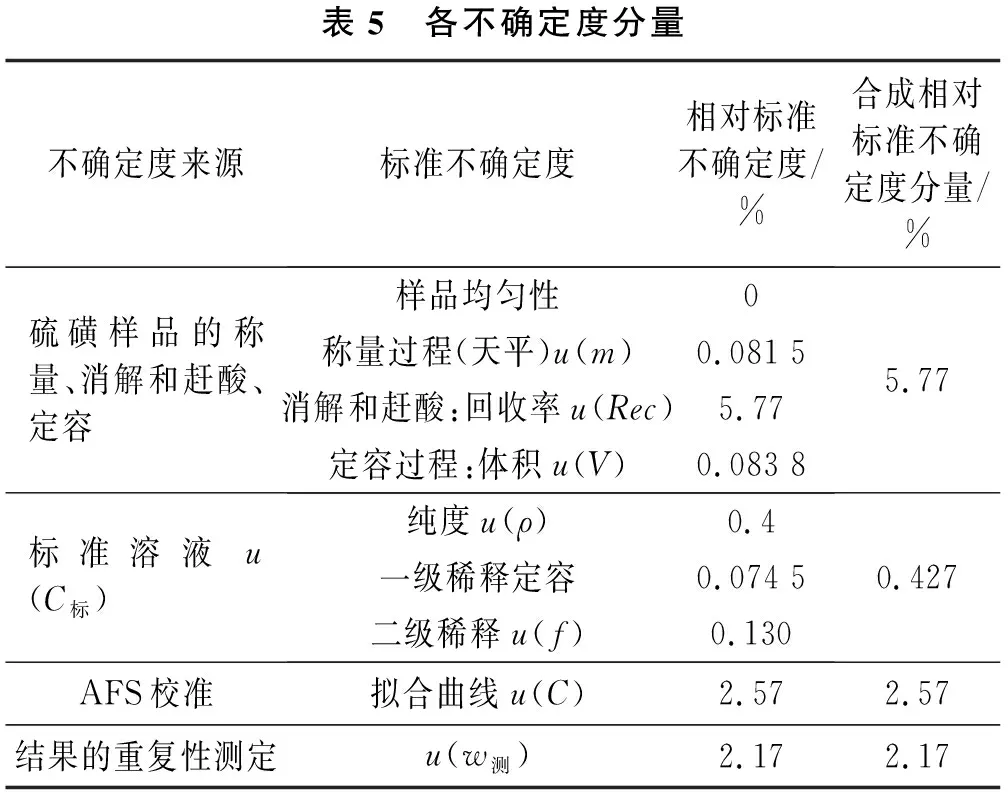
表5 各不确定度分量不确定度来源标准不确定度相对标准不确定度/%合成相对标准不确定度分量/%硫磺样品的称量、消解和赶酸、定容样品均匀性0称量过程(天平)u(m)0.081 5消解和赶酸:回收率u(Rec)5.77定容过程:体积u(V)0.083 85.77标准溶液u(C标)纯度u(ρ)0.4一级稀释定容0.074 5二级稀释u(f)0.1300.427AFS校准拟合曲线u(C)2.572.57结果的重复性测定u(w测)2.172.17
本方法的不确定度由样品处理、标准溶液、AFS校准和测量重复性引入的不确定度组成,其合成相对不确定度为:
urel(w)=
=6.69%
2.5 合成标准不确定度
合成标准不确定度由样品含量和合成相对不确定度计算,即:
u(w)=307 ng/g×6.69%=20.5 ng/g
2.6 计算扩展不确定度
扩展不确定度是由合成标准不确定度计算,与包含概率有关。采用的包含概率为95%,包含因子为2,扩展不确定度计算公式如下:
U=2u=20.5 ng/g×2=41 ng/g
2.7 测量不确定度
硫磺中As含量的测定结果为:(307±41)ng/g(k=2)
3 方法应用
采用本方法测定天然气产出硫磺中As含量测定,测量结果见表6。试验结果表明:天然气处理厂产出硫磺中As含量在(几十~几百)ng/g之间,小于1 μg /g,为优级品,即硫磺中的As含量均低于工业硫磺GB/T 2449.1-2014规定的优级品的要求。
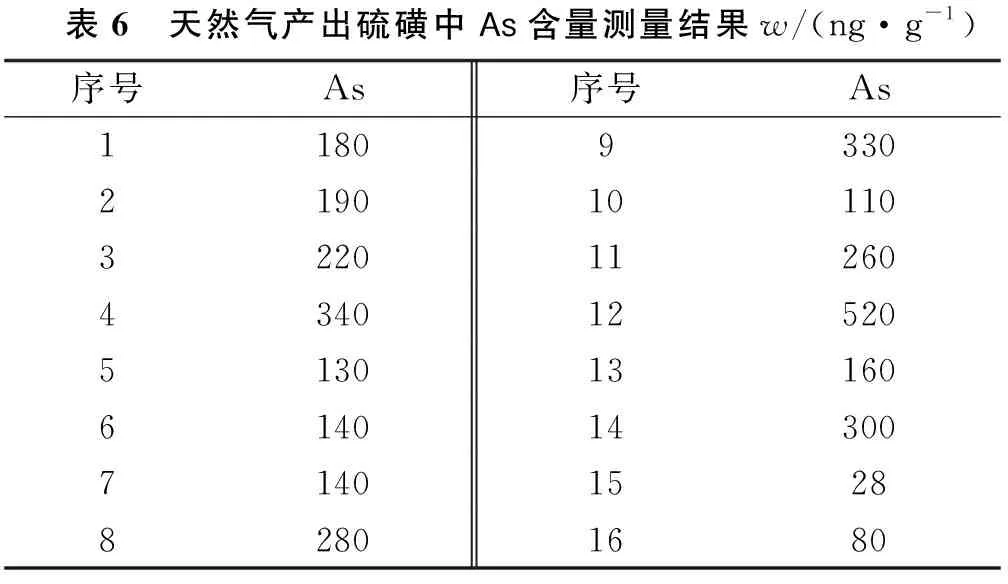
表6 天然气产出硫磺中As含量测量结果w/(ng·g-1)序号As序号As118093302190101103220112604340125205130131606140143007140152882801680
按第2.3节对表6中高、低两个检测结果其不确定度进行评定,高浓度不确定度贡献最大是消解回收率,为5.77%,低浓度不确定度贡献最大是标准曲线拟合,为26%,其测量结果不确定度:(520±66)ng/g(k=2)和(28±15)ng/g(k=2)。
4 结论
微波消解-原子荧光光谱法是准确测量硫磺中As含量的一种快速、环保、安全的方法,适用于天然气产出硫磺中微量As含量的测定。实验结果表明:天然气处理厂产出硫磺中As含量在(几十~几百)ng/g之间,小于1 μg /g,为优级品。微波消解-原子荧光光谱法测定硫磺中微量As含量的不确定度是由样品称量、样品消解、样品定容、标准溶液纯度、标准溶液稀释、标准曲线拟合和样品测量重复性等带来的。其中,消解回收率、标准曲线拟合和测量重复性是影响该方法不确定度的主要因素,对高、中浓度段不确定度贡献最大的是消解回收率,而对低浓度段则是标准曲线拟合。因此,在测量过程中应严格控制这3个方面的影响,降低各不确定度分量,提高测量结果的可靠性。
