Updates on “Cancer Genomics and Epigenomics”
2020-04-12AartiSharmaKiranLataSharmaCherryBansalAshokKumar
Aarti Sharma, Kiran Lata Sharma, Cherry Bansal, Ashok Kumar
Aarti Sharma, Ashok Kumar, Department of Surgical Gastroenterology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow 226014, India
Kiran Lata Sharma, Department of Pathology, Baylor College of Medicine, Houston, TX 77030,United States
Cherry Bansal, Department of Pathology, Era's Medical College and Hospital, Lucknow 226003, India
Abstract The field of “Cancer Genomics and Epigenomes” has been widely investigated for their involvement in cancer to understand the basic processes of different malignancies. The aggregation of genetic and epigenetic alterations also displays a wide range of heterogeneity making it quite necessary to develop personalized treatment strategies. The complex interplay between DNA methylation and chromatin dynamics in malignant cells is one of the major epigenetic mechanisms that lead to gene activation and repression. Hence, each tumor needs to be fully characterized to satisfy the ideas of personalized treatment strategies. The present article addresses various aspects of genome characterization methods and their potential role in the field of cancer genomics and epigenomics.
Key Words: Genetics; Epigenetics; Chromatin; Cancer; Copy number variations;Transcription; Histone-modifications
INTRODUCTION
"Cancer Genomics and Epigenomes" has played a critical role in our understanding of the basic processes of different malignancies. Multistep tumorigenesis is a progression of events, which result from the dysregulation of signaling mechanisms and alterations in the processing of genetic information. Cancer is a heterogeneous genetic disease caused by alterations in the genes that control cell growth and division. The major changes involve mutations in our genetic materiali.e.deoxyribonucleic acid(DNA) which makes our genes. The risk of developing cancer increases if genetic changes are inherited as germline changes and present in the germ cells. These changes are present in the entire cells of the progeny. Cancer-causing genetic changes can also be acquired during a person's life because of the errors in DNA repair mechanisms, which are caused by chemicals in tobacco, smoke, and radiation like ultraviolet rays. The genetic changes, which happen after conception at any time during the life of a person, are called somatic or acquired changes. The tumor cells can undergo various genetic changes like chromosomal rearrangements or genetic mutations.
Epigenetics is defined as heritable changes in gene activity that occur without alterations in the DNA sequence. These changes in gene expression are stable between cell divisions[1]. Some of these non-genetic variations are strongly controlled by two main epigenetic mechanisms,i.e.chemical modifications to the cytosine residues of DNA called DNA methylation and histone post-translational modifications associated with DNA. The complex interplay between DNA methylation and chromatin dynamics in malignant cells is one of the major epigenetic mechanisms that lead to gene activation and repression. These modifications play critical roles in the epigenetic inheritance of transcriptional memory. Complex patterns of epigenetic modifications can act as epigenetic markers to characterize gene expression, gene activity, and chromatin state[2,3]. Most epigenetic alterations are the global regulator of gene expression, affect the properties and behavior of the cells, and severely affect cancer progression.
The Cancer Genome Atlas, a database of comprehensive, multi-dimensional maps of the crucial genomic changes in 33 different cancers, that has been developed by a collaboration between the National Cancer Institute and National Human Genome Research Institute, comprises 2.5 petabytes data describing paired tumors and normal tissues of more than 11,000 patients. This database is freely available and is widely used by researchers (https://cancergenome.nih.gov/). The cBio Cancer Genomics Portal is a web-based (http://www.cbioportal.org/) open-access interactive platform of cancer genomics multidimensional datasets, which offer access to 5000 tumor samples from 20 various cancer studies. These efforts have lowered the walls between big genomic multifarious data and cancer researchers to translate the important information into biologic visions with applications to the clinic[4,5]. Nevertheless, the online cancer databases have recently evolved as a portal for integrative oncogenomics that stores data regarding gene information, microRNA (miRNA), protein expression profiling, copy number variations for several cancer forms, as well as protein-protein interaction information (http://www.canevolve.org/). It allows interrogating the consequences of the primary analysis, integrative analysis as well as network analysis of oncogenomics data[6]. A recent study published by Agarwalet al[7]created a Colorectal cancer Database, which contains the information from 2056 colorectal cancer (CRC)-related genes associated with different CRC stages, obtained from available literature with current information. This database is helping the clinicians obtain a better understanding of diagnosis as well as treatment and classification of CRCs http://lms.snu.edu.in/corecg/[7].
Next-generation sequencing (NGS)-based platforms such as the Ion Torrent PGM sequencer, MiSeq, HiSeq, reduced representation bisulfite sequencing, chromatin immunoprecipitation sequencing, methylation sequencing, methylated DNA immunoprecipitation, RNASeq, and array-based techniques are recently in use and have greatly assisted the discovery of biomarkers for the early diagnosis, prognosis,responses to chemotherapy, and treatment strategies in various cancers. In addition,the establishment of advanced NGS methodologies for less explored domains of cancer genomics such as non-coding RNA (ncRNA), miRNA, long ncRNA, small nucleolar RNA, and circular RNA (circRNA) have also revolutionized cancer research.
The present article addresses various aspects of genome characterization methods and their potentials role in the field of cancer genomics and epigenomics.
SINGLE-CELL SEQUENCING IN CANCER
The biological functions of cells are attained from transcription and translation.However, errors in their process can give rise to many genetic issues. Differences in gene regulation, stochastic variation, or environmental perturbations, are reflected at the genomic, transcriptomic, and proteomic levels. Cellular heterogeneity among cells within the same tumor is a result of genetic changes influenced by the environment.The heterogeneous nature of tumors is the primary reason for complicated treatment outcomes since a treatment that targets one tumor cell population may not be effective against another. To understand the role of rare cells in tumor progression single-cell sequencing is an emerging technology. Single-cell sequencing is a strong tool for investigating clonal evolution and heterogeneousness in cancer. With the help of currently available technologies for single-cell sequencing, it has become quite possible to characterize the intra-tumor cellular heterogeneity, measure mutation rates, and identify rare cell types that give rise to cellular heterogeneity. The hypothesis behind the development of carcinoma is the process of clonal evolution from mutated cells.Currently available technologies to identify rare cell typese.g., microfluidic-based single-cell sorting methods for stem cells, mass cytometry-based proteomic strategies,and high-throughput multiplexed quantitative polymerase chain reaction or sequencing methodologies. These methods provided new possibilities to look into the dynamical processes of cell-fate transitions[8].
Single-cell analysis in cancer genomics is highly important and new research orientation these days. Literature shows a good number of studies that have tried to explore the single-cell analysis approach to characterize cellular heterogeneity especially in the field of cancer genetics[9,10]. Bartoscheket al[10]studied the spatially and functionally distinct subclasses of breast cancer-associated fibroblasts by a single-cell RNA sequencing approach and found that there was an improved resolution of the widely defined cancer-associated fibroblast (CAF) population. The authors concluded that the single-cell RNA sequencing approach paves the way for the development of biomarker-related drugs directly for precision targeting of CAFs. Moreover, a recent review article by Macaulayet al[11]concluded that both genomic and transcriptomic heterogeneity within an organism has been considerably underestimated, and singlecell approaches now stand poised to illuminate this new layer of biological complexity during normal development and disease[11].
Exploring the field of cellular non-uniformity by single-cell sequencing approach will be very useful to impart fundamental insights into biological processes. Besides, it will also have important applications in the development of cancer-related therapies which is till now challenged by inter/intra heterogeneity.
COPY NUMBER VARIATIONS IN CANCER
Human genomes were once thought to be stable but current literature is full of publications with opposite views. An unexpectedly frequent, dynamic, and complex form of genetic diversity called copy number variations (CNVs) is an example of the dynamic nature of the genome. CNVs are a class of structurally variant regions within two human genomes. These discoveries have suddenly reversed the idea of a single diploid “reference genome.” The DNA CNVs are an important constituent of genetic variation, affecting a larger fraction of the genome as compared to single nucleotide variation. CNVs are greater than 1 kb base in size and defined as gains/losses of genomic DNA, which are of microscopic or submicroscopic level, therefore, not necessarily visible by standard G-banding karyotyping. The figures from March 2009 until the date of the Database of Genomic Variants have ascertained approximately 21000 CNVs or around 6500 unique CNV loci. The CNVs are believed to cover around 10% of the human genome. The cutting-edge technologies (NGS and microarray) have greatly facilitated the study of various aspects of human genomes. The recent advancements in medical research have made it possible to characterize the widespread constitutional CNVs, which have highlighted their role in susceptibility to a wide spectrum of diseases. The role of CNVs in cancer is currently underestimated and less understood. The CNV data can be used to identify regions of the genome involved in disease phenotypes because it affects the larger fraction of the genome.The advent of high-resolution technologies has made it possible to identify genomewide CNVs in comparatively less amount of time and are actively trying to determine the clinical impact of CNVs in patient populations. In particular, presence of CNVs leads to gene dosage variations and large-scale genome-wide investigations have demonstrated the large impact of CNVs on severe developmental disorders such as Down, Prader Willi, Angelman, and Cri du Chat syndromes, which result from gain or loss of one copy of a chromosome or chromosomal region. In cancer, copy number changes have also been involved in altered expression of tumor-suppressor as well as oncogenes. Thus, detection and mapping of copy number abnormalities provide significant information about the link of copy number aberrations with the disease phenotype, which ultimately helps the researchers in localizing critical genes.
TUMOR GENOTYPING IN CANCER
The development of targeted therapies for cancer patients greatly depends on tumor genotyping. A collaboration of pathologists and molecular biologists with clinical practitioners has shed a light on better, cost-effective, and comparatively faster methods to "genotype" malignant tumors to understand the genetic architecture of many cancers. Identifying the genetic abnormalities that drive particular tumors is leading to significant improvement in treatment-related modalities. It is evident from current data that genetic mutation based on targeted therapies work much better than conventional treatment methods. Moreover, personalized therapies can be common for two or more different cancers, irrespective of their tissue of origin.
MICROSATELLITE INSTABILITY AND CANCER
Genetic instability is a characteristic of most known malignancies, which involves a higher rate of mutations in the genome of cellular lineage. Genetic instability pathways are known to underlie mechanisms behind the development pattern of CRC. However,existing data concerning microsatellite instability (MSI) in hereditary colorectal cancer(85%) shows a well-marked distinction compared to sporadic CRC (up to 20%). It may be of particular interest that inactivated DNA mismatch repair (MMR) apparatus plays a crucial role in MSI occurrence, as loss or altered function of MMR proteins causes more error-prone DNA replication leading to the sequential accumulation of mutations.
MiRNA AND CANCER
miRNA or small ncRNA regularizes various target genes and hence engages in a variety of biological and pathological processes, like cancer. The role of miRNA in cancer has been discovered in several studies. Recent studies have identified mir-139-5p as a novel serum biomarker for CRC progression recurrence and metastasis[12].CircRNA “ciRS-7-A” is a potential therapeutic prognostic biomarker for CRC[13]. A study from the Yang group identified some novel approaches for studying circRNA functions. The authors found that circ-Foxo3 regulates the cellular functions of tissue cells and suppresses cancer cell progression in breast cancer[14]. The long ncRNA, H19,is significantly associated with CRC patient survival[15]. H19 silencing blocks the G1-S transition, inhibits cell migration, and reduces cell proliferation[15]. Regulation of Let-7[16], RB1-E2F1, and activity of β-catenin are also crucial upstream regulators mediating H19 function. Moreover, these ultra-conserved elements (i.e.miRNA) signify a newly recognized class of ncRNA, which assist in regulating miRNAs by direct interaction.These ultra-conserved genes expression signatures can help in better classification of tumors, including CRCs[17].
Cancer pharmacogenomics targets the understanding of how genetic variants influence drug efficacy and toxicity in cancer patient treatments. The inter-individual genetic variation affects a drug's pharmacokinetics, pharmacodynamics, and response to treatment[18]. As every patient responds to drug treatment differently, the genotypephenotype association can decide the drug dose. Pharmacogenomics-based databases have been developed in recent years for identifying patient target genes and small molecule candidates for cancer therapeutics[17]. A review article published on “cancer pharmacogenomics” in Nature genetics 2013 explains about various strategies and challenges including recent developments in sequencing techniques, clinical trial designs, and application of germline genetics analysis with bio-statistical genetics analysis methods that have potential for the detection of genetic variants associated with inter-individual response to the drug[19].
The list of genome/epigenome characterization techniques with their respective advantages and disadvantages is available in Table 1[20-30].
LIQUID BIOPSIES
Cells undergoing apoptosis or necrosis shed cell-free fragments of DNA into the bloodstream, which gives rise to circulating cell-free DNA. Recently, the technology has enabled to detect low levels of such a minor amount of cell-free DNA. In cancer,the load of circulating free DNA increases as the cells are increasingly undergoing cell division. The DNA shed by cancerous cells is known as circulating tumor DNA(ctDNA). ct-DNA contains the genetic information about the tumor and it has been evident from previous studies that ctDNA can also depict a wide range of clinical information like tumor genotyping, tumor staging, tumor grading, and prognosis,etc[31-34]. There are two major methods to study ct-DNA: One is by targeting ctDNA to find particular gene mutations/structural rearrangements in specific genome regions and the other one is the detection ofde novoctDNA mutations and somatic CNVs.These approaches are known as targeted and non-targeted approaches, respectively.The untargeted approach does not require any prior knowledge of molecular alteration, for instance, whole-genome sequencing. Both of these approaches have advantages and limitations. Genotyping ctDNA can prove a useful tool to detect any malignancy at the initial stage as well as the effect of ongoing treatment on patients without the involvement of invasive techniques. Table 2 lists the available CTC isolation methods[35-39].
CONCLUSION
In a nutshell, mutations, genome imbalances, and disruption of epigenetic machinery cumulatively characterize cancer. This evolutionary procedure for cancer progression requires interplay between the Cancer Genome and Epigenome. The newly formed malignant traits stably accumulate in the clonal lineage of cancer cells. Oncogenic events like mutation, copy number alteration, deletion, and insertion are particularly well recognized in the field of cancer biology. Cancer has been primarily studied based on genetics only. However, epigenetics turned out to be an alternative way of cancer that plays a significant role in acquiring stable oncogenic traits. Genetic and epigenetic mechanisms have a combined role in the formation of oncogenic traits by working in a multitude of ways as a team. Therefore, tumor characterization must include epigenomics along with genomics to get more accurate insights about the molecular pathology of the disease.
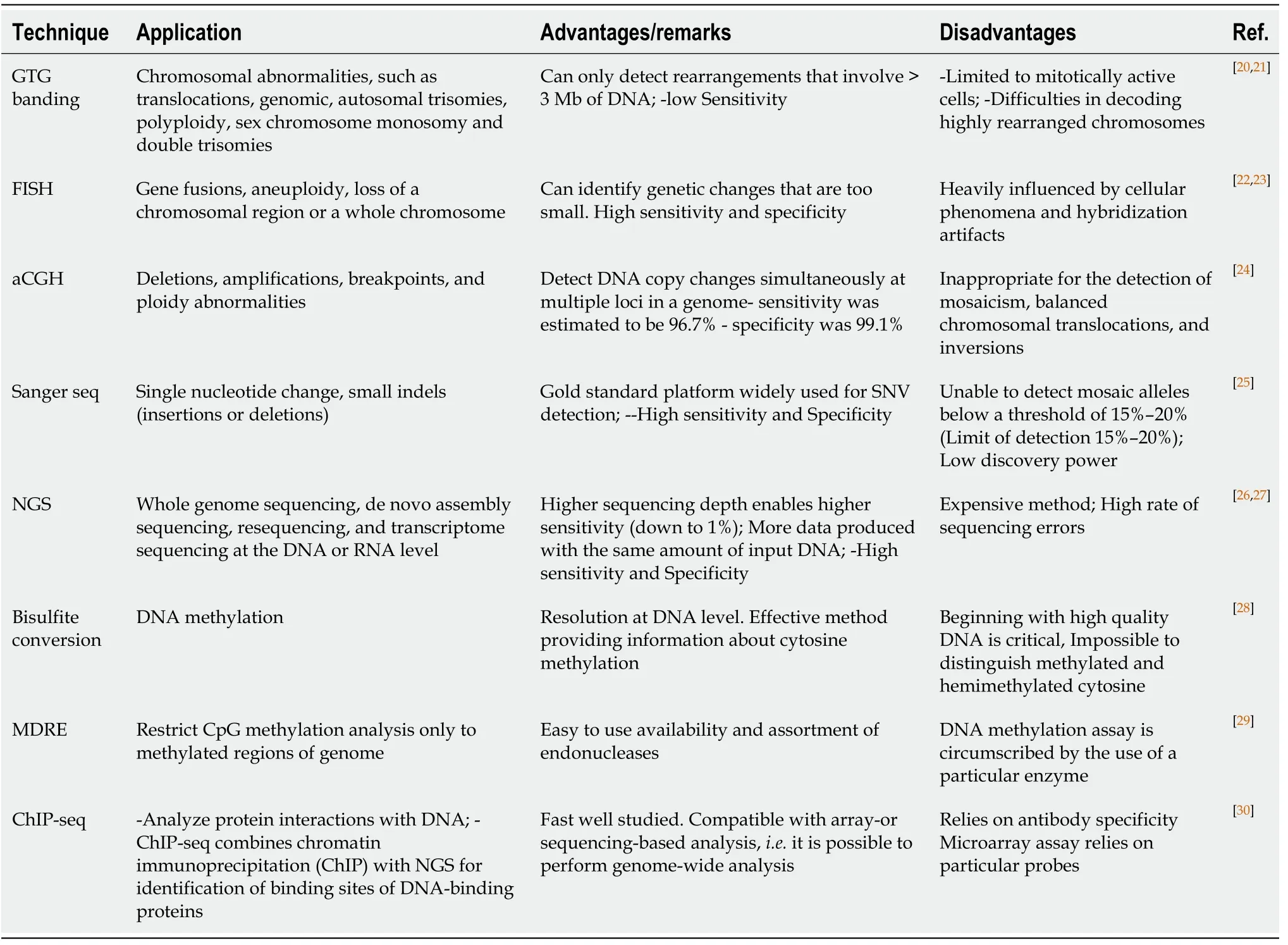
Table 1 Genome characterization technologies

Table 2 Circulating tumor cell isolation techniques
杂志排行

World Journal of Clinical Oncology的其它文章
- Tumor-specific lytic path “hyperploid progression mediated death”:Resolving side effects through targeting retinoblastoma or p53 mutant
- Liquid biopsy in ovarian cancer: Catching the silent killer before it strikes
- Deep diving in the PACIFIC: Practical issues in stage III non-small cell lung cancer to avoid shipwreck
- Artificial intelligence in dentistry: Harnessing big data to predict oral cancer survival
- Assessment of breast cancer immunohistochemistry and tumor characteristics in Nigeria
- Functional Gait Assessment scale in the rehabilitation of patients after vestibular tumor surgery in an acute hospital