Liquid biopsy in ovarian cancer: Catching the silent killer before it strikes
2020-04-12LauraFeeneyIanJGHarleyGlennMcCluggagePaulMullanJamesBeirne
Laura Feeney, Ian JG Harley, W Glenn McCluggage, Paul B Mullan, James P Beirne
Laura Feeney, Paul B Mullan, Patrick G Johnston Centre for Cancer Research, Queens University, Belfast BT9 7AE, United Kingdom
Ian JG Harley, Northern Ireland Gynaecological Cancer Centre, Belfast Health and Social Care Trust, Belfast BT9 7AB, United Kingdom
W Glenn McCluggage, Department of Pathology, Belfast Health and Social Care Trust, Belfast BT12 6BL, United Kingdom
James P Beirne, Trinity St James Cancer Institute, St. James' Hospital, Dublin 8, Ireland
Abstract Epithelial ovarian cancer (EOC) is the most lethal gynaecological malignancy in the western world. The majority of women presenting with the disease are asymptomatic and it has been dubbed the “silent killer”. To date there is no effective minimally invasive method of stratifying those with the disease or screening for the disease in the general population. Recent molecular and pathological discoveries, along with the advancement of scientific technology,means there is a real possibility of having disease-specific liquid biopsies available within the clinical environment in the near future. In this review we discuss these discoveries, particularly in relation to the most common and aggressive form of EOC, and their role in making this possibility a reality.
Key Words: Epithelial ovarian cancer; Molecular profile; Liquid biopsy; Circulating tumor DNA; Biomarker discovery; Precision medicine
INTRODUCTION
Epithelial ovarian cancer (EOC) is the most lethal gynaecological malignancy in the western world. In 2012 there were 152000 and 4300 deaths from ovarian cancer worldwide and in the United Kingdom, respectively[1]. This equates to twelve women dying from ovarian cancer within the United Kingdom every day.
The majority of women presenting with EOC are asymptomatic. In those women that do experience symptoms they are often vague and non-specific. The nature of the symptoms means that women generally present to their doctor with advanced stage disease. This has led to ovarian cancer being termed the “silent killer”. There have been major publicity campaigns, both regionally and nationally, involving the Department of Health and cancer charities to increase the level of awareness of ovarian cancer symptomatology; in an effort to improve earlier diagnosis.
EOC - FIVE DISTINCT DISEASES
There are five main types of EOC and these are included in the current World health Organization (WHO) classification: High-grade serous carcinoma (HGSC, 70%),endometrioid carcinoma (EC, 10%), clear cell carcinoma (CCC, 10%), mucinous carcinomas (MC, 3%), and low-grade serous carcinomas (LGSC, 3%)[2]. HGSC accounts for approximately 70% of all EOCs and approximately 90% of advanced stage EOCs(FIGO stage III-IV), making it the most common and most deadly.
The EOC types differ significantly morphologically, clinically, and at a molecular level. LGSC exhibitKRAS, BRAF,andERBB2mutations in around two thirds of cases whereasTP53mutations are very rare in these tumours[3,4].CTNNB1(encoding βcatenin) andPTENmutations, along with microsatellite instability, are associated with low-grade ECsviaspecific signalling pathways[5,6]. MC displayKRASmutations in more than 50% of specimens and identical mutations have been elucidated in benign,borderline and malignant areas from the same neoplastic lesion suggesting that aKRASmutation is an early event in mucinous tumour pathogenesis[7]. EC and CCC have been shown to be associated with endometriosis in the ovary or pelvis in 15% to 70% of cases; this is almost certainly an underestimate since endometriosis may be overgrown by the tumour and it is likely that a large majority of EC and CCC arise in endometriosis[8,9]. In fact, there is recent evidence to show that long interspersed element-1 hypomethylation is an early molecular event involved in the transformation of EC and CCC from endometriosis[10]. The tumour suppressor gene, AT-Rich Interaction Domain 1A (ARID1A) also seems to play a role in early malignant transformation of endometriosis to EC or CCC[11-13]. Mutations in this gene have been identified in up to 50% of CCCs and approximately one third of ECs[12].
HGSC differs significantly from the other subtypes. The classical histological appearance is of intermediate sized tumour cells, marked nuclear atypia, and necrotic areas (Figure 1). Immunostaining with WT1, PAX8, P16, and P53 assist with the diagnosis. Interestingly, WT1 staining helps discriminate between HGSC and pseudoendometrioid. Molecularly, it is characterised by the ubiquitous presence ofTP53mutations andCCNE1gene (encoding cyclin E1) amplification in 20% of cases[14-19]. It is, however, rarely associated with mutations such asKRAS,BRAF,ERBB2,HER2,PTEN,CTNNB1,ARID1AandPIK3CA[6,7,14,15,16,19]. Germline mutations in theBRCA1orBRCA2genes are present in 6.5%-19% of HGSCs and a smaller proportion have somatic mutations[20]. WhilstBRCA1somatic mutations are rare in sporadic disease (<10%), BRCA1 is reported to be downregulated in 15%-72%viamechanisms of epigenetic inactivation[20]. To complicate things further, the HGSC subtype is also highly molecularly heterogeneous, with the possible inclusion of further subpopulations that display distinct gene expression profiles and variable responses to current chemotherapy regimens[21].
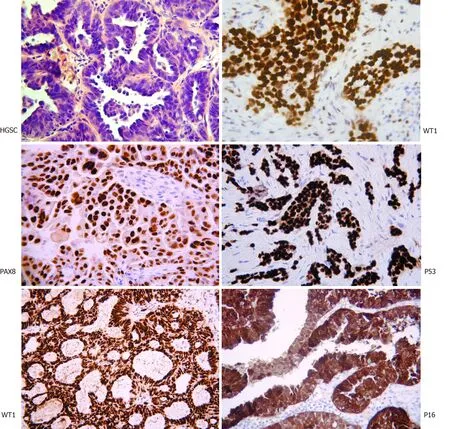
Figure 1 Histopathological assessment of high-grade serous carcinoma. The classical appearance on hematoxylin and eosin with intermediate sized tumor cells, marked nuclear atypia, and necrotic areas. Immunostaining with WT1, PAX8, P16, and P53 assist with the diagnosis. Interestingly WT1 staining helps discriminate between high-grade serous carcinoma and pseudo-endometrioid (bottom left) (Original figure, images courtesy of Professor McCluggage WG).
BLOOD-BASED DIAGNOSTICS OF EOC
The diagnosis of women with symptoms suspicious of EOC utilises the biomarker serum cancer antigen 125 (CA125), which was first described by Bastet al[22]in 1981. It was identified by the murine monoclonal antibody OC-125 as an antigenic determinant on a high molecular-weight glycoprotein. In adults, CA125 is expressed in tissues derived from both coelomic and Mullerian epithelia. It is also expressed by epithelia of the pancreas, colon, gall bladder, lung, kidney, and stomach[23].
In clinical practice, serum CA125 and preliminary radiological imaging, in the form of an abdomino-pelvic ultrasound, results are used to calculate the risk of malignancy index as a method of triaging patients for tertiary/quaternary referral[24]. Although it is used to aid diagnosis, only 50% of early stage EOCs show expression[23,25].
CA125 is most effective as a marker of disease status in patients undergoing chemotherapy treatment for EOC[26,27]. Unfortunately, it is not specific to malignancy,expressed by several other benign conditions including diverticulitis, endometriosis,liver cirrhosis, uterine fibroids, menstruation, pregnancy, pelvic infection, and uterine leiomyomata[28-33]. It is also elevated by other malignancies such as pancreatic, bladder,breast, liver, and lung cancers[33].
With earlier diagnosis the key to improved survival from EOC, there has been considerable effort to identify alternative biomarkers or develop combination markers with CA125, without overwhelming success[34]. To improve the sensitivity of CA125 the Risk of Ovarian Cancer Algorithm (ROCA) was developed[35]. This algorithm compares the CA125 level of cases with a profile of healthy women. It calculates a risk estimate based on this comparison and the age-specific incidence of EOC. The algorithm was employed within the UKCTOCS trial and various others with varying degrees of success[36-38].
The only novel biomarker that has showed promise, since CA125, is human epididymis protein 4 (HE4)[39]. This biomarker has been shown to improve diagnostic accuracy, both alone and in combination with CA125[29,40-42]. A recent meta-analysis looked at diagnostic performance with two control groups (healthy women and women with benign gynaecological disease)[40]. In the analysisvs“healthy women” the sensitivity and specificity for HE4 in diagnosing ovarian cancer were 83% (95%CI:77%-88%) and 90% (95%CI: 87%-92%), respectively. Receiver operator characteristic(ROC) analysis revealed an area under the curve (AUC) of 0.9271. In the women with benign disease analysis, the sensitivity and specificity were 74% (95%CI: 69%-78%) and 90% (95%CI: 87%-92%) respectively. The ROC analysis AUC was 0.8853. This suggests HE4 carries potential as an early-warning biomarker. Contrastingly, another metaanalysis looking at CA125 and HE4 combined diagnostic performance showed HE4 to be no better than CA125 for predicting EOC[43].
A panel of biomarkers that included CA125, HE4, transthyretin, CA15.3, and CA72.4 was evaluated using specimens assembled from multiple cohort and randomised trial[44]. Phase II and III biomarker studies concluded that CA125 remained the “single-best biomarker” for EOC. Another retrospective study evaluated seven proteomic biomarkers (apolipoprotein A1, truncated transthyretin, transferrin,hepcidin, beta-2 microglobulin, connective tissue activating protein III, and interalpha-trypsin inhibitor heavy-chain) in pre-diagnostic blood samples[45]. The addition of the seven protein biomarkers to CA125 did not improve sensitivity compared to CA125 alone.
The combination of CA125 and HE4 was developed into an algorithm in an effort to improve EOC detection[46]. The Risk of Ovarian Malignancy Algorithm (ROMA)successfully classified 93.8% of EOC patients as high risk. The model has been assessed in a range of populations with varying degrees of accuracy[42,47-53]. It is for this reason that HE4 or ROMA has not translated into routine clinical care, with some groups requesting further in-depth validation.
CATCHING THE SILENT KILLER: ATTEMPTS TO DATE
The evidence that population screening impacts cancer-specific mortality is everaccumulating. There are examples of successful cancer screening programmes across many countries, particularly for breast, bowel, and cervical cancers. A significant reduction in mortality has been seen in the latter[54]. The real success of these screening programmes is the understanding of the pathogenesis of the disease. Currently there is no effective screening method for ovarian cancer and the United States Preventative Services Task Force (USPSTF) maintains its position that it should not be undertaken,in any form[55].
Ovarian cancer is relatively rare with a low prevalence in the general population.An effective and acceptable screening strategy must have not only a high sensitivity for early-stage disease (> 75%) but must also have a very high specificity (99.6%) to achieve a positive predictive value (PPV) of 10%. In real terms, that equates to a threshold of no more than 10 staging laparotomies to identify one ovarian cancer[56].
The main methods of ovarian cancer screening assessed to date are pelvic ultrasound scanning and serially measuring the serum biomarker, CA125. Pelvic ultrasound in expert hands is a highly sensitive diagnostic method[57]. Unfortunately,because it relies heavily on individual expertise, discrimination between benign and malignant pelvic masses in routine clinical practice is challenging. Serum CA125 is most effective as a marker of disease status in patients undergoing chemotherapy treatment for EOC[26]. It is not particularly specific to malignancy and can be expressed by a number of other benign conditions including endometriosis, pelvic infection, and uterine leiomyomata. It is elevated in only 50% of early stage EOCs, and thus can cause unnecessary medical intervention and significant patient distress[25].
There have been a few large prospective trials examining ovarian cancer screening.Each utilised different screening strategies but all aimed to assess the same primary outcome: Mortality.
The United States Prostate Lung Colorectal Ovarian Cancer Screening Trial(USPLCO) comprised over 78000 women aged 55-74 years in two arms; annual transvaginal ultrasound and serum CA125vsroutine clinical care[58]. The study showed no difference in mortality between the two cohorts. The United Kingdom Collaborative Trial in Ovarian Cancer Screening (UKCTOCS) is the largest screening trial to date, enrolling over 200000 postmenopausal women randomised into no intervention or quarterly (initially annual) screening[38]. The screening cohort was broken down into transvaginal ultrasound alone or serum CA125 screening,interpreted by the ROCA algorithm, with second-line transvaginal ultrasound (termed multimodal screening). UKCTOCS finally reported mortality data in 2016. At a median follow-up of 11.1 years, ovarian cancer was diagnosed in 1282 (0.6%) women. There was no difference across subgroups with 0.7% in the multimodal screening group,0.6% in the ultrasound only group, and 0.6% in the control group. Of these women,0.29% in the MMS group, 0.30% in the USS group, and 0.34% in the control group died of ovarian cancer. The primary analysis revealed a non-statistically significant mortality reduction of 15% (P= 0.10) with MMS and 11% (P= 0.21) with USS alone.
Although the primary end point did not reach statistical significance, further analysis suggested improved mortality reductions for the MMS group compared to the control group after 7 years; 8% reduction for the first 7 years compared to 23% for years 7 to 14; suggesting a “late effect” mortality benefit[38]. In response to the “late effect” mortality trend proposed by the UKCTOCS study, extended mortality results were reported for the USPLCO trial with a median 15 year follow up (range 13 to 19 years)[59]. However, following this extended analysis, the study reiterated its initial findings and reported no change in the mortality benefit of screening using CA125 and TVUS.
NEW PATHOLOGICAL KNOWLEDGE
To date there have been no disease-specific biomarkers validated for any of the five main EOC types. Moving forward, this approach would be more appropriate given that these represent different tumour types with a different pathogenesis and behaviour and require different management. Currently, the spotlight is on HGSC as it is the most frequent and most aggressive type of EOC.
For some time, scientists and clinicians have been unable to identify a pre-invasive stage to HGSC, and the disease did not appear to fit this model. This is likely explained by the fact the disease spreads so aggressively quite early in its course,making the pathological detection of early stage disease elusive[60]. There has been considerable effort employed to define the molecular mechanisms of HGSC and, until recently, its pathogenesis remained undefined. Historically, it was thought HGSC developed from the ovarian surface epithelium (OSE) due to errors in cell replication associated with the repair of trauma incurred by ovulation[61]. Several epidemiological studies supported this theory with evidence that women with an increased number of lifetime ovulations are at a much greater risk of developing HGSC[62-67].
However, in recent years, overwhelming pathological evidence has emerged that supports the theory that the distal fallopian tube (fimbria) is the origin of HGSC[68]. The fact that fallopian tube epithelium is embryologically mullerian and HGSCs are mullerian in nature suggests a potential site of origin here for HGSC. Furthermore,malignant transformation of fallopian tubal epithelium yields almost exclusively HGSC[69]. HGSC occurring concurrently with fallopian tubal mucosal disease was first documented by Bannatyneet al[70]but this was interpreted to represent a second primary tumour focus rather than being directly related to the ovarian disease.Subsequently a number of research groups acknowledged that there was an increased risk of primary fallopian tubal HGSC inBRCA1/2mutation carriers[71-73]. Zweemeret al[74]presented the first evidence of primary tubal HGSC inBRCA1mutants and,subsequently, a large prospective study ofBRCA1mutation carriers revealed a 120-fold increased risk, compared to the general population, of primary fallopian tube carcinoma[75]. In 2006, Finchet al[76]published clinical and pathological findings of prophylactic salpingo-oophorectomy specimens from 159BRCA1/2mutation carriers.Seven (4.4%) occult fallopian tube cancers were identified in these women, in the absence of symptoms. Multiple other investigators also recorded the presence of occult tubal tumours at prophylactic salpingoophorectomy[76-83]. The incidence of these occult lesions ranged from 6%-40% with up to 100% of them occurring in the fallopian tube,usually in the absence of ovarian involvement. Further investigation has revealed most HGSCs arise from the distal fallopian tubeviaan in-situ carcinomatous lesion referred to as serous tubal intraepithelial carcinoma (STIC)[84,85].
THE FALLOPIAN TUBE AS THE ORIGIN OF HGSC
There is now compelling evidence from both our group and others worldwide that most, but not all, HSGCs arise from the distal fallopian tube, and not the ovary, from STIC[86-88]. Detailed assessment of the FTs in cases of extrauterine HGSC shows involvement of the fimbriae in 70% and STIC in approximately 50% of the cases[89]. At a molecular level; Cyclin E1 (CCNE1), remodelling and spacing factor 1 (Rsf-1), and fatty acid synthase (FASN) are all upregulated in HGSC. Rsf-1 encodes an important ATP-dependent chromatin remodelling protein which is an integral component of DNA replication and cell cycle progression[90,91]. FASN is a cytoplasmic enzyme involved in tumour initiation and progression[92,93]. These oncogenes are all overexpressed in STIC lesions[94,95]. Another oncogene integral to this disease process isTP53[96]. Mutation of this tumour suppressor is known to be associated with HGSC and recent evidence shows it be characteristic of HGSC and essentially ubiquitous[15,97,98].The fact that STIC lesions and concurrent HGSC have been shown to contain identicalTP53mutations further indicates a clonal relationship between the two entities[98,99].Gene expression profiling of a unique sample set containing normal OSE, normal FT,STIC, ovarian HGSC, and omental metastases was performed by our group.Bioinformatic analysis revealed that the tumour samples clustered in one cohort and normal FT samples clustered together and separately from the normal OSE samples.Notably, the normal OSE samples clustered separately from all other profiled samples and STIC samples clustered within the tumour cohort. Multi-dimensional scaling analysis confirmed the strong common biology present between STIC and the two tumour groups. It also affirmed that OSE has no significant common genetic biology to the other samples. This study further confirms the fallopian tubal origin of HGSC[86].
The proportion of HGSCs derived from the FT is currently unknown, mainly due to the fact HGSC usually presents at an advanced stage making the detection of precursor lesions difficult and depends on the criteria used to determine a tubal primary. However, in the most detailed prospective study, using criteria for site assignment of extrauterine HGSC adopted by the International Collaboration on Cancer Reporting (ICCR), 83% of cases were determined to be of tubal origin and almost all the remainder of ovarian origin with a primary peritoneal origin being extremely rare when strict criteria are used[100-103].
EXPLOITING THIS NOVEL MOLECULAR KNOWLEDGE: THE LIQUID BIOPSY
Precision oncology seeks to obtain molecular information about cancer to improve patient outcomes. Tissue biopsy samples are widely used to characterise tumours;however, this method of tumour analysis has limitations. The term “liquid biopsy”was first used to describe methods that can derive the same diagnostic information from a blood sample, or other body fluid, that is typically derived from a tissue biopsy sample[104]. In recent years, the focus of precision medicine is increasingly turning towards liquid biopsies as they are minimally invasive and can be repeated at multiple time points facilitating “real-time” disease monitoring[105].
Liquid biopsy (Figure 2) can include measurement of soluble factors, such as circulating tumour nucleic acids (DNA/RNA), circulating tumour cells (CTCs),proteins, and extracellular vesicles such as exosomes. All of which have been investigated for potential as diagnostic, predictive and prognostic biomarkers[106].
Figure 2 The difference between liquid and traditional tissue biopsy (Original figure).
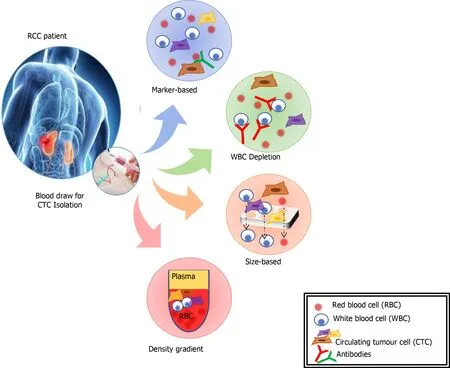
Figure 3 The extraction of circulating tumor cells can be undertaken by a range of methods (reproduced from Broncy et al[166] 2018 under CC BY-NC 4.0). RCC: Renal cell carcinoma.
CTCs are cells originating from a solid tumour that are detectable in the peripheral blood. They are considered a prerequisite step in establishing distant metastases[107].The detection of CTCs in peripheral blood (Figure 3) is a novel type of cancer biomarker[108]. CTCs can be isolated from blood samples and used to follow patients over time. They can provide significant information that will better characterise underlying disease biology and metastases. However, CTCs are rare, and their isolation, quantification and molecular characterization carry many challenges. An average metastatic carcinoma patient has between 5 and 50 CTCs for every 7.5 mL of blood[109]. This places technical limitations on the ability to identify and characterise the sub-population of cells that carry the relevant genetic information. CTCs tend to aggregate with leucocytes so adequate cell-surface markers and separation techniques need to be utilised to improve purity[109].
CTCs were first detected on the background of malignant melanoma and have subsequently been identified associated with a range of solid tumours[110]. There are a range of approaches for isolation of CTCs including; mRNA-based, protein-based, and cell size-based. The mRNA-based strategy involves RqPCR techniques. This carries some limitations including: (1) Amplification of non-specific products, (2) Lack of validated protocols for sample processing, RNA-preparation, cDNA synthesis and PCR conditions, and (3) A lack of sample quality control measures[107]. These issues all raise the possibility of variations in sensitivity and specificity of a biomarker employing this method. Separation and enrichment of CTCs, using magnetic bead technologies, followed by flow cytometry or immunohistochemistry is another method of quantification and characterisation of CTCs[106]. This method eliminates some of the concerns with RqPCR techniques, but further research needs to be done to clarify the reproducibility of different techniques. The isolation by size of epithelial tumour cells is a direct method for CTC identification and has been applied in various epithelial cancers[107]. The CTCs are collected by filtration and, following staining for specific markers, the cells are identified and quantified by immunohistochemistry or molecular pathological techniques.
The only clinically validated, FDA-approved blood test for CTCs is the CELLSEARCH®CTC Test (Janssen Diagnostics, New Jersey, United States). It has shown promise as a prognostic indicator in prostate, colorectal, and breast cancers[111-113]. This technology was assessed within the EOC population and while it did provide evidence that ovarian CTCs were present in blood it did not correlate with clinical outcomes[113]. A further study assessed a unique cell adhesion matrix based,functional cell enrichment and identification platform[114]. There was clear evidence that elevated CTCs correlated with worse OS and PFS. In fact, this method proved more specific than CA125 in detecting EOC malignancy in high-risk patients. A recent meta-analysis assessed eleven studies comprising a total of 1129 patients[115]. It reported CTC status to be a significant prognostic indicator (OS:HR 1.61, 95%CI: 1.22-2.13; PFS:HR 1.44, 95%CI: 1.18-1.75). A subgroup analysis showed the RqPCR methodology to be superior to both CELLSEARCH®and immunohistochemical methods.
Despite the potential role of CTCs in cancer diagnostics, CTC methods are generally used for research purposes, and only a few methods have been accepted for clinical application. This is because of the difficulties caused by CTC heterogeneity, CTC separation from blood, and a lack of thorough clinical validation[116].
In recent years there has been a significant amount of research into the use of circulating cell free DNA (cfDNA) as a biomarker. The presence of circulatory cfDNA was identified over seventy years ago[117]. Along with technological evolution,subsequent research has demonstrated that cancer cells release cfDNA fragments into the circulation and other bodily fluids, termed circulating tumour DNA (ctDNA), and these fragments carry all the genetic and epigenetic characteristics of the primary tumour[118]. Fragments of cfDNA in blood samples are between 150 and 200 bp long, of which up to 90% originates from the tumour[118]. It is reported that tumours containing approximately 50 million malignant cells release enough DNA for the detection of tumour cfDNA in blood[119]. Of note, this is well below the limit of resolution of radiological imaging (approximately 1 billion cells)[120].
As a result, ctDNA analysis has emerged as a potential blood-based “liquid biopsy”for early detection, diagnosis, staging and prognosis, monitoring response to treatment, monitoring minimal residual disease and relapse and identifying acquired drug resistance mechanisms.
There are several hypotheses as to the origin and mechanism of release of cfDNA/ctDNA into the circulation; however, the precise mechanism(s) have yet to be determined. Early studies suggested that ctDNA enters the circulation following lysis of cells on the interface between the tumour and circulation[121]. Another theory proposed that ctDNA may originate from the destruction of tumour micro metastases and circulating cancer cells[122]. Current consensus suggests that most cfDNA in healthy individuals is released from the bone marrow and white blood cells, whereas ctDNA in cancer patients is derived from necrotic and apoptotic cancer cells. Three possible mechanisms resulting in the shedding of DNA from both healthy and tumour cells have been described: Apoptosis, necrosis, and active cellular release[123]. Apoptosis causes the systematic cleavage of chromosomal DNA into multiples of 160-180 bp stretches, resulting in the extracellular presence of mono-(approximately 166 bp) and poly-nucleosomes (332 bp, 498 bp)[124]. Necrosis results in nuclear chromatin clumping and non-specific digestion, producing DNA fragments that are typically larger than 10000 bp. DNA fragments derived from active cellular secretions have been shown to range between 1000 and 3000 bp. Most of the DNA present in plasma occurs as fragments around 180 bases and 360 bases in size and reflects the likely apoptotic origin of the DNA[125].
In cancer patients, cfDNA may originate from multiple sources, including cancer cells, cells from the tumour microenvironment and normal cells such as haematopoietic stem cells, muscle cells and epithelial cells. Once present in circulation,cfDNA levels are influenced by multiple factors including its: (1) Dynamic association and disassociation with extracellular vesicles and several serum proteins, (2) Rate of binding, dissociation and internalisation by cells; and (3) Rate of digestion or clearance, including the activity of deoxyribonuclease I (DNAse), renal excretion into urine, and uptake by the liver and spleen[126]. Furthermore, cfDNA levels can be elevated due to the lysis of white blood cells (WBCs) and release of germline DNA,thus diluting ctDNA concentrations[127]. For this reason, it is crucial that during molecular analysis tumour-specific ctDNA is differentiated from non-tumour cfDNA and contamination of blood samples through WBC lysis is kept to a minimum.
THE LIQUID BIOPSY IN CLINICAL PRACTICE
Disease staging
Studies have demonstrated that cfDNA levels in cancer patients are generally higher than those of healthy subjects. cfDNA is present in healthy individuals at average concentrations of 30 ng/mL, ranging from 0-100 ng/mL[118]. In cancer patients, given the additional release of cfDNA from tumour cells, the average concentration of cfDNA is much higher, at approximately 180 ng/mL[128]. cfDNA concentration has also been shown to correlate with tumour size, disease stage and metastatic burden[129-131].
In a study of 640 patients with different cancer types at varying stages, Bettegowdaet al[131]showed that cfDNA levels were approximately 100 times higher in stage IV disease compared to stage I disease, providing a rough estimate of tumour size based on cfDNA concentration. In another study, aimed at providing a more sensitive metric for estimating tumour size, researchers reported that mutant alleles increased by 6 mutant copies per ml of plasma for every cubic centimetre of tumour in participants with HGSC[129].
In one of the first studies to examine cfDNA concentration in EOC, Kamatet al[130]demonstrated that cfDNA levels were elevated in advanced EOC compared to normal controls. In a more recent study, the same group evaluated the role of preoperative total plasma cfDNA levels in predicting clinical outcomes in patients with EOC[132].Again, they reported significantly higher cfDNA levels in the EOC group (median 10113 genomic equivalent/mL, GE/mL) compared with benign ovarian tumours(median, 2365 GE/mL;P< 0.001) and unaffected controls (median, 1912 GE/mL;P<0.001). Moreover, a statistically significant association of cfDNA > 22000 GE/mL with decreased PFS (P< 0.001) was observed, which was superior to CA125 in predicting mortality. Furthermore, the study reported elevated cfDNA levels in patients with early stage disease which was significantly higher compared with those with benign disease and controls (P< 0.01). Shaoet al[133]also reported significantly elevated cfDNA levels in advanced stage OC compared to early stage (P< 0.01). In contrast, Noet al[134]reported no significant difference between cfDNA levels in EOC patients compared to controls. In this study, preoperative blood samples of 36 EOC patients and 16 benign tumours were analysed using commercially available copy number assay kits to measure cfDNA levels of four genes; beta-2-microglobulin (B2M), member RAS oncogene family (RAB25), claudin 4 (CLDN4) and ATP-binding cassette subfamily F member 2 (ABCF2). This result may be explained, in part, by the fact that, unlike the previous studies, this study used serum instead of plasma as the cfDNA source.
In a study investigating the presence of tumour-specificTP53sequences in blood and peritoneal fluid in EOC patients, Swisheret al[135]detected 30% (21/69) of patients with confirmedTP53mutations (exon 2-11) in plasma or serum samples. cfDNA was detected in 93% (28/30) of cases in peritoneal fluid, including six cases with negative cytology. Following multivariate analysis, they concluded that detection of cfDNA was associated with decreased survival (P= 0.02). Dobrzyckaet al[136]investigated the prognostic significance of cfDNA and blood plasma p53 antibodies (p53-Ab) in EOC.Serum p53-Ab is predominantly associated withTP53gene missense mutations andTP53accumulation in the tumour. cfDNA and p53-Ab were more frequently detected in patients with HGSC (P< 0.001) compared to other EOC subtypes. Prognosis was significantly worse in cfDNA positive patients compared to cfDNA negative (P=0.022). Similarly, patients who were p53-Ab positive had a significantly worse prognosis than those who were p53-Ab negative (P< 0.001).
Whole exome sequencing (WES) and targeted gene sequencing has been employed to identify tumour-specific mutations in gynaecological cancers[137]. Subsequent monitoring of these mutations using ctDNA and digital PCR technology, in patients with gynaecological cancers, detected the recurrence of cancer, on average, seven months before disease was visible on cross-sectional imaging. Furthermore,undetectable levels of ctDNA at six months following initial treatment was associated with significantly improved progression free and overall survival.
The correlation between ctDNA levels and tumour stage/size highlights the potential prognostic value of ctDNA. Several studies have demonstrated an association between ctDNA and survival outcome in a range of cancers[138].
Personalised therapy
Assessing the mutational profile of cancer patients is used as a stratification tool to identify those who may be suitable for targeted therapies. For example, in patients with non-small cell lung cancer (NSCLC), the tyrosine kinase inhibitors (TKI) gefitinib and erlotinib are only beneficial to those with an activating mutation (L858R or exon 19 deletion) in the epidermal growth factor receptor (EGFR)gene[139]. Similarly,patients with malignant melanoma will only benefit fromBRAFtherapy if they harbour an activatingBRAFmutation (V600E)[140]. Identifying these mutations and others enables clinicians to alter therapies accordingly and optimise patient care.
Historically, the assessment of mutational status has been carried out using tissue biopsies, however, this practice has some inherent disadvantages. The analysis of a single tissue biopsy taken from a primary tumour or metastatic site is likely to underestimate the mutational landscape of a tumour and can lead to inaccurate classification. Performing several biopsies on one patient can be impractical and extremely invasive. Furthermore, tissue biopsies are associated with complications,such as infection and pain, and often fail to obtain enough material for high quality mutational profiling. Numerous studies, in various malignancies, have shown that these limitations may be overcome by mutational profiling of cfDNA[106].
In CRC, high concordance between tissue and plasma samples have been reported for the detection ofKRAS, NRASandBRAFmutations, with concordance rates of 91.8% reported in one study forRASmutations[141]. Another study suggested that cfDNA could replace tumour-tissue analysis resulting in considerable reductions in data turnaround time[142].
There are a limited number of studies investigating the role of cfDNA and treatment resistance in EOC. Murtazaet al[143]carried out WES in serial plasma samples to track the genomic evolution of metastatic cancers in response to treatment. Three patients with advanced EOC were included in the study. Quantification of allele fractions in plasma identified mutant alleles association with emerging treatment resistance. This study established a proof-of-principle that WES of ctDNA could complement current invasive biopsy approaches to identify mutations associated with acquired resistant in advanced cancers.
Overall, these studies demonstrate high concordance between tissue biopsies and plasma samples with higher diagnostic accuracy recorded using cfDNA analysis in most cases.
Treatment monitoring
The short half-life of cfDNA, coupled with the minimally invasive nature of venepuncture compared to tissue biopsies makes cfDNA an attractive tool for monitoring treatment response and disease burden (Figure 4). Numerous studies have demonstrated that low levels of cfDNA are associated with a positive treatment response in a range of cancers, including EOC[144]. In contrast, high levels of cfDNA generally correlate with poor response to treatment, treatment resistance, high risk of relapse and poor survival. In the majority of these studies, cfDNA was reported to monitor response to treatment more accurately than traditional methods. In one example, Capizziet al[145]investigated the role of cfDNA levels in predicting response to chemotherapy in EOC. In this prospective non-randomised clinical trial, 22 patients with advanced EOC (FIGO stage IIIC or IV) undergoing neo-adjuvant chemotherapy were recruited alongside 50 female healthy blood donor controls. Plasma cfDNA levels were quantified before, during and after chemotherapy. Median cfDNA levels were reported to be significantly higher in the cancer group (29.6 ± 22.7 ng/mL) prior to chemotherapy, compared to controls (6.4 ± 4.0 ng/mL), with a sensitivity of 77% and specificity of 96%. A general trend was found between elevated cfDNA levels on completion of chemotherapy and disease progression (P= 0.007), however, the sample size was too small to provide conclusive survival data.
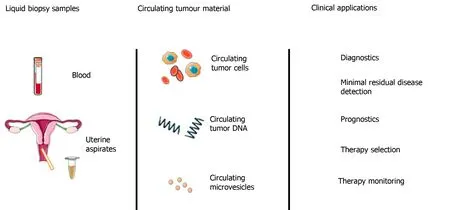
Figure 4 Potential liquid biopsy sources (blood, uterine/cervical aspirates) and downstream clinical applications within epithelial ovarian cancer (reproduced from Muinelo-Romay et al[167] 2018 under CC BY-NC 4.0).
In a retrospective study of 40 patients with relapsed HGSC, Parkinsonet al[129]used sequence specific assays to detect predefinedTP53mutations and quantified theTP53mutant allele frequency (TP53MAF) in cfDNA using digital PCR before, during and after chemotherapy. Pre-treatment ctDNATP53MAF concentration was positively correlated with total volume of disease, and a decrease of > 60% after one cycle of chemotherapy was associated with longer PFS.
Identifying treatment-resistant disease
Acquired drug resistance may emerge as a result ofde novomutations or the expansion of a sub-clonal cell population with pre-existing resistance[146]. The underlying mechanisms of acquired resistance are poorly understood. However, longitudinal sampling and analysis of cfDNA can provide valuable insight into the molecular response of cancer during treatment.
cfDNA can be used to monitor the development of resistance by screening for known mutations associated with resistance. Using a digital PCR assay, Ishiiet al[147]examined cfDNA in plasma from patients with relapsed NSCLC to identify resistance mutations, namely T790M mutations, associated with EGFR-TKIs. T760M mutation was detected in plasma with a sensitivity of 81.8% and specificity of 85.7%, and overall concordance between plasma and tissue samples was 83.3%. This study showed that digital PCR analysis of plasma is a feasible and accurate alternative to tissue biopsy for detecting T760M mutations in NSCLC patients that become resistant to EGFR-TKIs.
Serial profiling of cfDNA can identify resistant sub-clones before the onset of clinical progression and enable earlier intervention. In a study investigating the acquired resistance to anti-EGFR treatment in CRC, 60% (6/10) patients showed the emergence of secondaryKRASmutations up to four months before an increase in the conventional marker (CEA) was detected, and nine months prior to radiological evidence of relapse[148]. This study also showed that although tumour cells exhibited resistance to EGFR inhibitors, they remained sensitive to a combination of EGFR and MEK inhibitors, enabling early and personalised treatment adjustment.
A key resistance mechanism to platinum-based chemotherapies and PARP inhibitors inBRCA-mutant cancers is the acquisition ofBRCAreversion mutations that restore protein function. Linet al[149]performed targeted next-generation sequencing of cfDNA extracted from plasma collected prior to rucaparib treatment in 112 patients with germline or somaticBRCA-mutant HGSC enrolled in the ARIEL2 study. They foundBRCAreversion mutations in cfDNA from 18% (2/11) of platinum-refractory and 13% (5/38) of platinum-resistant patients, compared with 2% (1/48) of platinumsensitive patients (P= 0.049). Furthermore, patients withoutBRCAreversion mutations detected in pre-treatment cfDNA had significantly longer rucaparib PFS than those with reversion mutations (median, 9.0 movs18 mo; HR, 0.12;P< 0.0001).
In summary, analysis of cfDNA collected before and after treatment can provide a more comprehensive view of the genetic response of a patients' tumour, including the dynamic changes in the mutational landscape as well as the heterogeneity that develops due to the selective pressure of therapy.
Minimal residual disease
Another important aspect of cancer management is deciding whether further treatment is required following tumour resection. Decision-making for adjuvant therapy is based on disease stage and certain high-risk clinical or pathological features.As there is, currently, no effective tool of identifying patients with complete tumour resection this practice leads to potential under- and over-treatment with the associated consequences.
cfDNA analysis has shown great promise in the detection of minimal residual disease (MRD) and subsequent risk of recurrence. In one of the first studies to evaluate the use of cfDNA as a biomarker of MRD, Diehlet al[150]showed that tumour derived cfDNA levels decreased by 99% within 24 h after complete surgical resection in CRC,whereas in cases of incomplete resection cfDNA levels did not change significantly and in some cases increased. Furthermore, in subjects with detectable levels of cfDNA after surgery relapse generally occurred within one year. cfDNA levels appeared to be a more reliable and sensitive indicator than the conventional biomarker(carcinoembryonic antigen)in this cohort. More recently, Chaudhuriet al[151]demonstrated the potential of cfDNA deep sequencing analysis in predicting prognosis in patients who had completed potentially curative treatment for early-stage NSCLC with surgery or radical radiotherapy. They retrospectively analysed blood samples from a cohort of 40 patients, with plasma samples taken before treatment and every 2 to 6 mo during follow-up. ctDNA was detectable in 93% (37/40) of patients before any treatment and was detectable in 54% of patients after treatment, all of whom went on to relapse. Detection of ctDNA postoperatively had a very high risk of future relapse (HR, 43.4; 95%CI, 5.7-341), with a median 5.2-mo lead time over clinical progression. Further studies have reported similar findings in a range of malignancies,including EOC[126].
Although these studies are encouraging there remains a large proportion of patients who relapse in whom cfDNA is not detected. Furthermore, the vast majority of these cfDNA assays are patient and mutation specific, limiting their clinical application to wider patient populations. In order to overcome these limitations further research is required to establish highly sensitive cfDNA assays that enable the identification of patients likely to benefit from adjuvant treatments and avoid the adverse effects resulting from unnecessary treatments.
CATCHING THE SILENT KILLER: A BETTER WAY?
Despite years of research in this area, the diagnosis of early stage cancer remains extremely challenging. Recent research suggests that technological advances in the analysis of cfDNA may provide a solution to these challenges. Studies have shown that cancer-associated mutations can be detected in cfDNA in early-stage disease,before the presence of symptoms and up to 2 years before cancer diagnosis[152-156].
Cervical screening tests have revolutionised the management of cervical cancer by enabling early detection of preinvasive disease. Recently, the traditional Papanicolau smear has been replaced, in many countries, by a liquid-based cytology (LBC) method.Kindeet al[152]exploited this method of DNA collection to develop an assay to detect endometrial and ovarian cancer. Mutational profiling was carried out on 46 cancer patients (24 endometrial cancers and 22 ovarian cancers) for whom LBC specimens were available. The same mutations were detected in the corresponding LBC samples in 100% (24/24) of the endometrial cancers and 41% (9/22) of the ovarian cancers. The same group went on to develop the PapSEEK test using a similar technique to analysis a panel of 18 genes using multiplex PCR[157]. They reported sensitivity of 33% (95%CI,27%-39%) in the 245 EOC patients tested, including 34% of patients with early stage disease. Specificity was approximately 99% with only 1.4% of 714 women without cancer testing positive. They also analysed plasma in 83 EOC patients for 16 genes of interest and found ctDNA in 43% (95%CI, 33%-55%), with none of the plasma samples from 192 healthy controls testing positive (specificity 100%). When combining LBC samples with matched plasma samples, sensitivity for OC was increased to 63%(95%CI, 51%-73%), including 54% with early stage disease. Although improvements are required before applying this test in routine practice, it highlights the potential to incorporate cfDNA analysis into routine screening tools such as the cervical screening programme.
Non-invasive prenatal testing (NIPT) identifies foetal aneuploidy by sequencing cfDNA in maternal plasma. Pre-symptomatic maternal malignancies have been incidentally detected during NIPT[155]. In a case control study of prospectively collected preoperative HGSC plasma samples, Cohenet al[153]analysed 32 women with HGSC(16 early stage (FIGO I-II) and 16 advanced stage (FIGO III-IV)) and 32 benign controls.Plasma cfDNA was sequenced using a commercial NIPT platform. They detected 40.6% (13/32) HGSC cases using sub-chromosomal analysis, including 38% (6/16) of early stage cases. Although sensitivity was low and correlation with paired tumour DNA was not possible due to a lack of archived specimens, this study established the proof-of-principle that tumour DNA is detectable using NIPT.
The same group developed a blood test to detect eight common cancers through assessment of levels of circulating proteins and mutations in cfDNA[154]. The CancerSEEK®test was used to assess 1005 patients who had been diagnosed with stage I to III cancers of the ovary, liver, stomach, pancreas, oesophagus, colorectum, lung and breast. The median sensitivity of CancerSEEK®among the eight cancer types was 70% (P< 10-96) and ranged from 98% in OC to 33% in breast cancers. At this sensitivity,the specificity was > 99%; with 7 of the 812 controls recorded as positive. Although sensitivity was reported as > 70% for stage II (73%) and stage III (78%) disease, only 43% of stage I cancers were detected. A key concern with this test is its achievable PPV. The prevalence of the eight cancers in healthy individuals > 64 years of age is approximately 1%. Assuming the CancerSEEK®test could achieve 99% sensitivity and specificity, the resulting PPV would be only 50%, which equates to 50% of positive tests being false positives. However, these results imply that combination strategies have the power to greatly improve liquid biopsy analyses.
Gormallyet al[156]assessed the significance of plasma DNA mutations for subsequent cancer development in healthy subjects in a large longitudinal prospective study. The study included > 520000 healthy volunteers recruited from 10 European countries.Plasma specimens were tested forTP53andKRAS2mutations. Results showed that mutations inTP53andKRAS2could be detected in cfDNA of healthy subjects on average 20.8 mo (range, 1.8-44.8) and 14.3 mo (range, 2.6-24.9) before cancer diagnosis,respectively.TP53andKRAS2mutations were detected in 3% and 1%, respectively, of subjects who did not develop cancer during follow up. This is an important finding as it highlights the presence of high levels of oncogenic drivers in the plasma of healthy individuals. This has been attributed to a common aging-related phenomenon known as clonal haematopoiesis, in which haematopoietic stem cells or other early blood cell progenitors contribute to the formation of genetically distinct subpopulations of bloods cells[158]. The establishment of a clonal population may occur when a stem or progenitor cell acquires one or more somatic mutations that give it a competitive advantage over other haematopoietic cells. The incidence of clonal haematopoiesis rises with age and is an important consideration when evaluating potential bloodbased tumour-specific biomarkers.
Whilst these studies demonstrate the potential of cfDNA as an early detection marker, several significant obstacles need to be overcome before the majority could be used in a clinical setting. The main obstacles to the development of cfDNA based biomarkers are: (1) Low abundance of ctDNA in the blood; and (2) High levels of background non-cancerous cfDNA, mostly shed from WBCs. Highly sensitive technologies are required to accurately detect scarcely abundant alleles within high background levels of nontarget molecules.
Due to this, cfDNA-based cancer biomarkers have yet to make it into the clinical arena. The greatest progress has been observed in obstetric medicine, where cfDNA has been successfully used for fetal Rhesus D genotyping, for the detection of paternally inherited genetic disorders from maternal blood, and revolutionised the identification of fetal aneuploidy through non-invasive pre-natal screening[159]. In the future this level of success may be seen for cancer-specific cfDNA biomarkers.
CONCLUSION
The use of liquid biopsies is a fast-emerging area of cancer diagnostics, in particular the detection of ctDNA, and multiple cancer types are known to produce quantifiable levels of ctDNA. Studies have shown ctDNA to be useful in monitoring for minimal residual disease, treatment response, chemoresistance, and tumour heterogeneity.ctDNA can be used as an early warning diagnostic tool, either through identification of known genetic aberrations (such asTP53mutations), or through measurement of cancer-specific DNA methylation (DNAme) of particular genomic loci. However, use of this technology for early diagnosis is very much in its infancy. In EOC it has been shown thatTP53mutations can be identified in ctDNA, using tagged amplicon deep sequencing, in up to 65% of patients with advanced EOC[160]. Once optimised this could be a very useful liquid biopsy for HGSC but it would be important to ascertain theTP53mutation rate in normal healthy controls as a baseline.
At present, sequencing technology allows for the detection of one mutant allele,e.g.,p53, in a background of 1000 wild-type molecules[161]. For this reason, the current focus of our research is on DNAme because it allows for the detection of specific patterns rather than single point mutations, potentially improving the performance characteristics and detection limit of such an assay. There have been numerous reports of alterations in methylation events occurring in EOC, including HGSC[162-164]. This could prove to be an extremely useful mechanism through which HGSCs might be detected earlier and more consistently.
The identification of a biomarker requires a number of phases of development which can be crudely described as; case and control selection, determination of detection limits and assay precision, validation in second/external datasets, statistical interpretation and ROC analysis. The failure to find suitable biomarkers for EOC,despite significant investment in the United Kingdom, Europe and the United States,led to an inquiry into possible causes. This inquiry recommended the use of the PRoBE(prospective-specimen collection, with retrospective-blinded evaluation) design for biomarker discovery and validation because it was felt that biomarkers discovered in clinical sample sets collected at diagnosis from symptomatic patients and controls in hospital settings are unlikely to fully represent the screening population[165]. The PRoBE design involves blinded case-control studies nested within a prospective cohort representing the target population. The specimens and matched clinical data will have been collected prior to knowledge of the outcome (e.g., diagnosis of EOC). Paramount to the successful implementation of the PRoBE design is the knowledge of the accurate calibrator-control for the disease the biomarker is aimed for.
As discussed, the clinicopathological and molecular developments over the last decade has redefined EOC as essentially representing five distinct disease entities. It is now the time to consider identifying disease-specific biomarkers rather than a generic EOC tumour marker. This will likely yield more success and, ultimately, result in increased precision of the biomarker.
HGSC is the major contributor to morbidity and mortality under the EOC“umbrella”. Therefore, an accurate disease-specific biomarker is urgently needed; not just for “screening” but for greater diagnostic accuracy and also for monitoring disease stability/progression during treatment and assessing residual disease post cytoreductive surgery. Our use of cutting-edge, high throughput, molecular assay technologies has helped clarify the underlying molecular profile of this disease[86].Knowledge like this will guide the identification and validation of novel transcripts that carry the potential to act as disease-specific biomarkers of HGSC.
杂志排行

World Journal of Clinical Oncology的其它文章
- Tumor-specific lytic path “hyperploid progression mediated death”:Resolving side effects through targeting retinoblastoma or p53 mutant
- Updates on “Cancer Genomics and Epigenomics”
- Deep diving in the PACIFIC: Practical issues in stage III non-small cell lung cancer to avoid shipwreck
- Artificial intelligence in dentistry: Harnessing big data to predict oral cancer survival
- Assessment of breast cancer immunohistochemistry and tumor characteristics in Nigeria
- Functional Gait Assessment scale in the rehabilitation of patients after vestibular tumor surgery in an acute hospital