SiH(X2Π)从头算解析势能函数和光谱常数
2020-04-09赵文丽高峰张红曹学成
赵文丽,高峰,张红,曹学成
山东农业大学信息科学与工程学院,山东泰安 271018
SiH 自由基是许多化学反应(如硅烷分子裂解反应)的重要中间产物,它在诸多工业过程,如等离子气体沉积、薄膜构成以及半导体制造业方面有着重要的作用[1,2]。另外,SiH 自由基广泛存在于在星际物质[3]和恒星气体中(在太阳黑子的光谱中已得到证实[4,5]),其光谱数据已经成为天体物理学的重要参数。因此,不论是在天体物理学领域还是化学工程领域SiH 自由基都是重要的研究对象。
实验上,1930 年Jackson[6]首次采用光谱法观测到SiH 自由基。1931 年,Mulliken[7]对Jackson观测到的光谱进行了分析和部分修正。1980 年Perrin 和Delafosse[8]用硅烷辉光放电管测量了SiH 自由基发射光谱。1985 年Washida[9]在硅烷的真空紫外线光分解系统内观察到SiH 自由基(A2Δ→X2П)的发射光谱。1987 年Seebass[10]通过激光磁共振观测到了SiH 自由基5 个同位素的红外光谱,并且观察到了SiH 基态(X2П)的自旋和轨道耦合效应。1998 年Ram[11]用高分辨率的傅里叶变换光谱仪观测到了SiH 自由基的发射光谱。这些实验报道了SiH 自由基的光谱常数和分子特性。
理论上,1967 年Cade 和Huo[12]首次运用从头算方法给出SiH 自由基的光谱常数,并用Hartree-Fock 方法计算了SiH(X2П)的光谱常数。1971 年Wirsam[13]结合自洽场分子轨道和组态相互作用(CI)方法计算SiH 自由基六个态的光谱性质,其中三个态当时还没有实验数据报道。1983 年Lewerenz 和他的合作者[14]用多参考双激发组态相互作用(MRD-CI)方法计算了SiH 自由基10 个态的势能曲线(PECs)。施德恒等于2008 年采用单双体三激发耦合簇(CCSD(T))方法研究了SiH(X2П)的光谱常数,又于2013 年使用多参考组态相互作用(MRCI)方法计算了SiH(X2П,A2Δ,B2Σ-,C2Σ+,D2Σ+,F2П and a4Σ-)的势能曲线和光谱常数[15,16]。这些理论研究报道了SiH 自由基的部分态的光谱常数。
1 理论
1.1 从头计算
我们选用MRCI(Q)和戴维孙修正方法[17]进行从头算,用aug-cc-pVXZ(AVXZ)和aug-ccpV(X+d)Z(AVXdZ)(X=Q,5,6)的基组,由Molpro2012 软件包计算得到所有从头算能量点。采用C2v点群对称进行计算,其中包含四个不可约表示,即A1,A2,B1 和B2。计算中将9 个分子轨道(MO)确定为活化空间(4-8σ和2-3π轨道),他们包含5 个a1,2 个b1 和2 个b2 对称分子轨道。Si 原子在3S3P 轨道上的4 个电子和H 原子1S 轨道上的1 个电子被放在活化空间,即这5 个电子分布在上述9 个分子轨道上,剩下的10 个电子放在5 个闭壳层轨道,包括3 个a1,1 个b1,1 个b2,相当于分子的1-3σ和1π轨道。对于每个基组获得的PEC,核间距都选择在0.06 nm 到1.00 nm 之间。应该指出,分子单点能的计算都是使用有限基组来完成的,这必然产生基组重叠误差(BSSE)。Varandas[18]提出BSSE 可以通过将从头算能量点外推到完备基组(CBS)[19-23]极限来纠正。因此,我们选择CBS 的方法修正从头算能量点。
1.2 基组外推
根据Varandas 提出的基于Dunning 相关一致基组外推电子能量的方案[22],MRCI 电子能量可以写成:

其中R 为指定空间坐标的三维向量,下标表示能量是用AVXZ 基组计算的,上标CAS 和dc 分别表示完备活性空间能量和动态相关能量。CAS 能量使用Karton 和Martin[21]提出的双点外插方案,将AVQZ 和AV5Z 基组外推至CBS 极限。公式为:
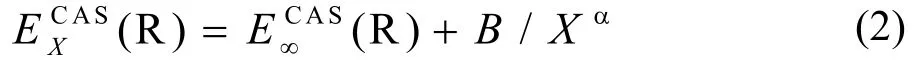
其中B为取决于从头算能量点的参量,α=5.34 为有效的衰变指数,(R)为X→∞的能量。dc 能量为:
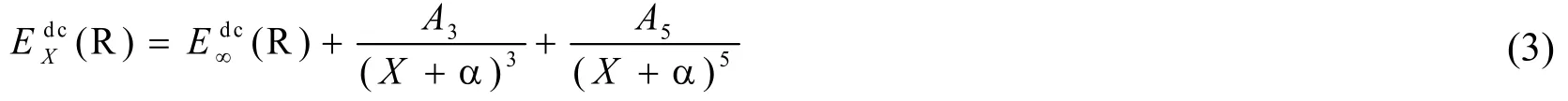
其中A5满足关系:
且A5(0)=0.0037685459Eh,Eh为能量的原子单位,1Eh=27.2116 eV,c=-1.17847713Eh-1/4,α=-3/8,和A3由AVQZ 和AV5Z 基组拟合dc 能量决定。
1.3 解析势能函数(APEFs)
SiH(X2П)APEFs 采用Aguado 和Paniagua[24]提出的函数形式:

Vshort和Vlong分别为短程和长程相互作用的势能,其中:
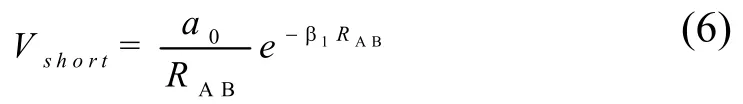

式(6)表明当RAB→0 双原子相互作用势趋近于无穷,式(7)当RAB→∞双原子相互作用势趋于零。
解析势能函数分别通过拟合外插到CBS 极限的数据(使用AVQZ 和AV5Z 的计算数据)和AV6Z的计算数据得到。最后采用最小二乘法拟合程序得出线性参数αi(i=0,1,2,…,n)和非线性参数βi(i=1,2)。
2 结果和讨论
2.1 势能函数
我们分别使用几种不同的基组(AVQZ,AV5Z,AV6Z,USTE(Q,5)),获得SiH(X2П)的能量点,从这些能量点出发,应用(5)式拟合得到APEFs,为了增加精确度,使用了12 个参数,得到了几种不同基组下的APEFs,函数曲线的参数列于表1。
图1 是SiH(X2П)在AV6Z 基组和CBS 方法(使用AV(Q,5)Z 基组)拟合得到的PECs,圆点表示从头算能量点,实线是拟合得到的PECs,从图中可以看出,两种方法得到的PECs 都表现出平滑和收敛的行为,且在AV6Z 和USTE(Q,5)下拟合的PECs 与从头算能量点之间的误差小于5 cm-1,二者之间吻合得非常好。
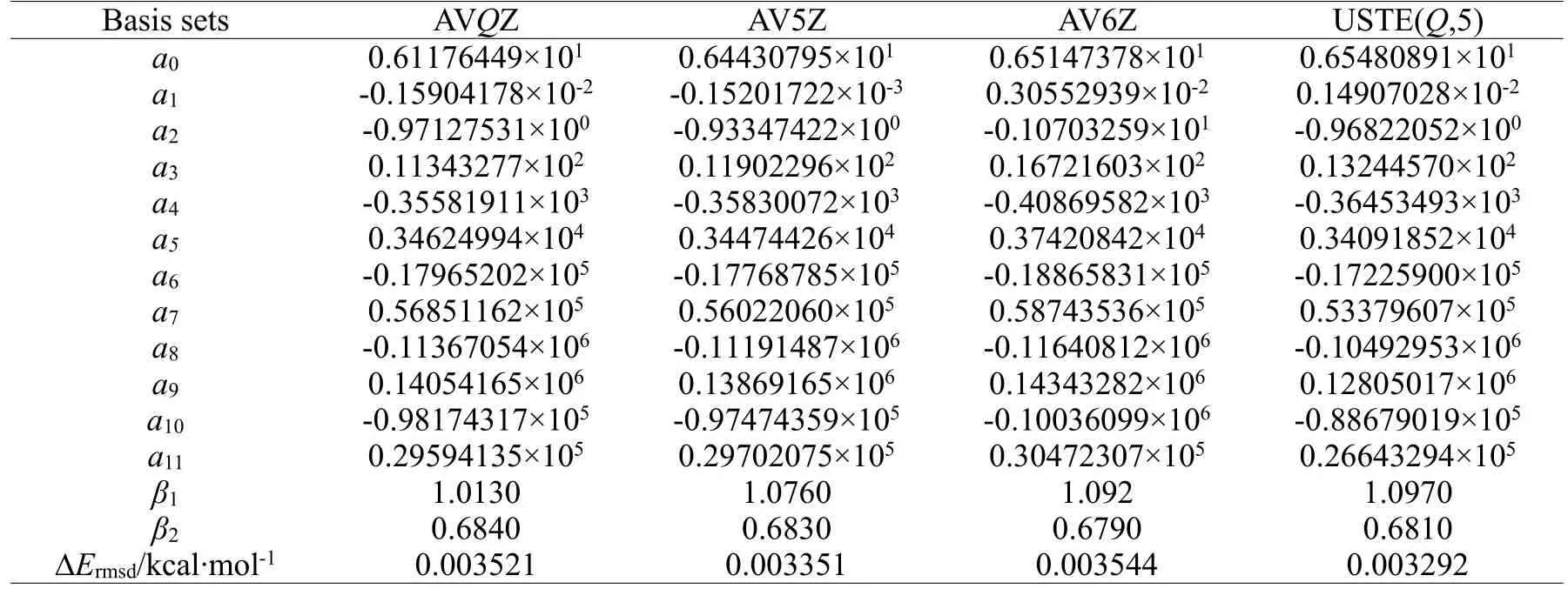
表1 拟合SiH(X2Π)APEFs 的参数Table 1 Fitting the parameters of SiH(X2Π)APEFs

图1 SiH(X2Π)的势能曲线Fig.1 PECs for SiH(X2Π)
为了评估PECs 的拟合质量,可以计算SiH(X2П)在不同基组下的均方根误差(rmsd),理论公式为:

其中,Vfit(i)和Vab(i)分别是第i个拟合能量和从头算能量,N是拟合能量点的个数,取N=92。ΔErmsd也在表1 中列出。从表中可以发现,对于X2П 态MRCI(Q)/AVQZ,MRCI(Q)/AV5Z,MRCI(Q)/AV6Z 和CBS/USTE(Q,5)的ΔErmsd分别为0.003521 kcal/mol,0.003351 kcal/mol,0.003544 kcal/mol 和0.003292 kcal/mol。使用USTE(Q,5)方法得到的势能函数曲线与从头算能量点之间的误差比使用AV6Z 基组更小,ΔErmsd从0.003544 减小到0.003292 kcal∙mol-1。目前普遍认为AV6Z 是一个耗时量极大的基组,其计算成本远高于AVQZ 和AV5Z。通过使用USTE(Q,5)外推方案,可以在显著降低计算成本的同时得到准确的势能曲线。
2.2 光谱常数
运用拟合MRCI(Q)/AVQZ,MRCI(Q)/AV5Z,MRCI(Q)/AV6Z 和CBS/USTE(Q,5)势能曲线获得的APEFs,计算出SiH(X2П)的光谱常数,以及其他一些高质量的理论和实验研究结果都列在表2中。其中,前四行为不同基组计算的结果,第5,6 行为实验结果[25,26]。7~12 行依次是一些其他文献计算结果[12,16,27-30]。如Cade[12]等人通过求解H-F-R 方程的数值解得到了平衡键长Re,解离能De和αe。但与实验值相比,他们的振动频率ωe过大,解离能De、αe、ωeχe则偏小。总体来看,若以实验数据为参考,Kalemos 等人的理论计算结果[29]是最精确的。另外,从表中前四行可以看出,De随基组从AVQZ 到AV6Z 是单调增加的,其最大值是由CBS/USTE(Q,5)APEF 获得,与AV6Z APEF 获得的De差值为0.00796 eV。由CBS/USTE(Q,5)APEF 计算出的Re为1.5221a0,比两个实验值长0.0020a0和0.0024a0,比Kalemos 等人获得的最新值仅相差0.0002a0,显示出较高精度。CBS/USTE(Q,5)APEF 计算的ωe为2043.26 cm-1,这与实验[26]和理论[29]值分别相差0.74 cm-1和0.11 cm-1。CBS/USTE(Q,5)APEF 计算的光谱常数βe小于实验值,这是因为βe与Re成反比。另外,光谱常数αe与实验[26]和理论[29]值也吻合得非常好,然而,CBS/USTE(Q,5)APEF 计算的ωeχe却和实验值偏差较大,这是因为ωeχe受βe,ωe和力常数(二次方f2,立方f3和四次方f4)的影响,所有这些因素都可能导致ωeχe的值大于实验值。

表2 SiH(X2П)双原子平衡键长Re,解离能De,振动频率ωe,光谱常数ωeχe,αe,βeTable 2 Diatomic equilibrium bond length Re,dissociation energy De,vibrational frequency ωe,and other spectral constant ωeχe,αe,βeof SiH(X2 П)
2.3 振动能级
核运动的径向薛定谔方程为:
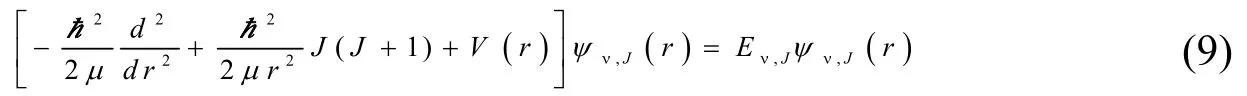
其中ψν,J(r)和Eν,J分别是的本征函数和本征值,V(r)是APEF 的能量,r是双原子SiH 的核间距,μ是分子的约化质量,v和J分别是振动量子数和转动量子数。对于给定的振动能级,转动能级可以写成:

其中G(v)是振动能级,Bv是转动常数,Dv,Hv,Lv,Mv,Nv和Ov是离心畸变常数。
方程(9)可以通过Level7.5 程序包求解,通过解方程可以获得SiH(X2П)的振动能级。进而得到当转动量子数J=0 时的SiH(X2П)的振动态。表3 列出了前21 个振动态的结果。对于每个振动态,获得了G(v)及其经典拐点,一个转动常数Bv和六个离心畸变常数Dv,Hv,Lv,Mv,Nv和Ov。

表3 SiH(X2П)当J=0 时的21 个振动能级G(v)(cm-1),经典拐点和转动常数Bv(cm-1)Table 3 21 vibration levels G(v)(cm-1),classical turning point and rotation constants Bv(cm-1)of SiH(X2П)when J=0
表3 给出了由CBS/ USTE(Q,5)和AV6Z APEFs 计算的SiH振动能级G(v),经典拐点及其转动常数Bv。当v=0 时,两种方法计算得到的G(v)差值仅为0.814 cm-1,随着振动量子数v的增加,差值变大,当v=20 时,最大差值为59.470 cm-1。在v=20 处发现Bv的最大差值,其值为2.60×10-2cm-1。此外,经典拐点差值在10-3a0的幅度内,两种方法计算结果表现出一致性。图2 是根据表3 CBS/USTE(Q,5)数据画出的SiH(X2П)振动能级图。
基于CBS/USTE(Q,5)计算的转动量子数J=0 时SiH(X2∏)前21 个振动态的结果列于表4 中。由于目前没有发现关于SiH(X2∏)振动能级、经典拐点、转动常数和离心畸变常数的实验或其他理论工作报告。因此,这些理论数据没有可以比较的对象。但是,根据高质量拟合的APEFs,以及目前计算的光谱参数与其他文献结果的一致性[12,16,25-30],我们认为表3 和表4 中收集的结果是可靠的。
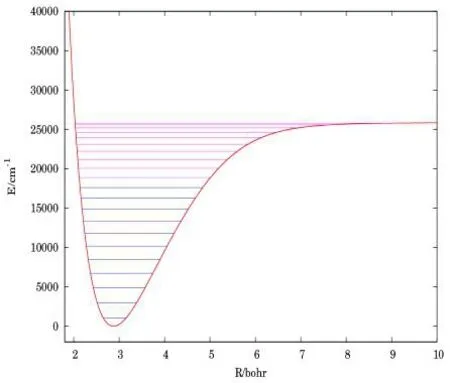
图2 SiH(X2П)振动能级图Fig.2 Vibration energy level of SiH(X2П)
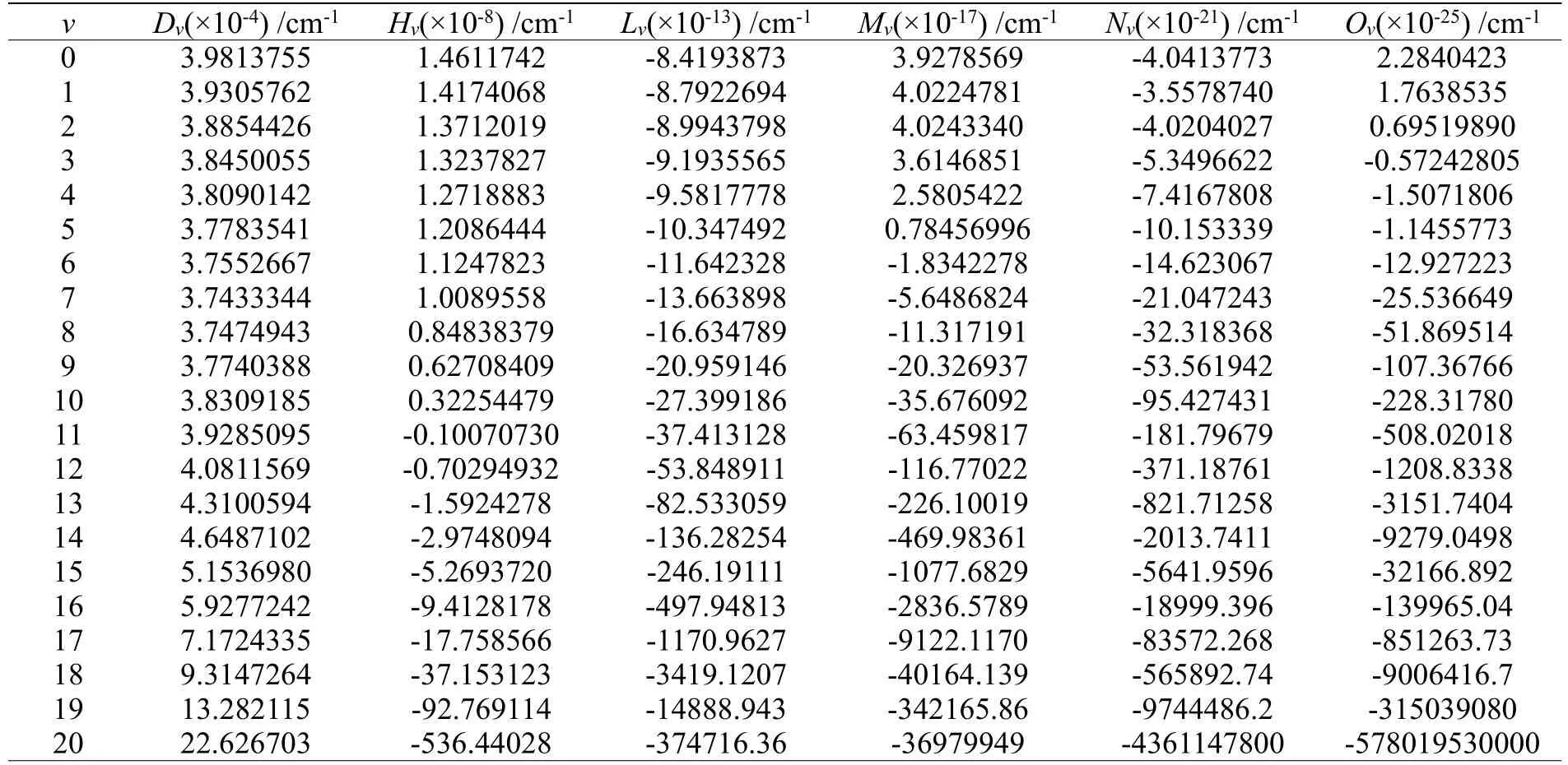
表4 由CBS/USTE(Q,5)APEFs 计算的SiH(X2П)(J=0)的21 个离心畸变常数(cm-1)Table 4 21centrifugal distortion constants(cm-1)of SiH(X2П)(J=0) calculated by CBS/USTE(Q,5)APEFs
3 结论
基于MRCI(Q)方法和aug-cc-pVXZ(AVXZ)和aug-cc-pV(X+d)Z(AVXdZ) (X=Q,5,6)基组,得到了SiH(X2П)从头算能量点,然后采用USTE(Q,5)方案将势能曲线能量点外推到CBS 极限。通过能量点拟合获得APEFs。基于APEFs 我们用不同方法计算了光谱常数De,Re,ωe,Be,αe和ωeχe。通过与已有的实验值和其他理论结果相比较,发现用CBS/USTE(Q,5)和CBS/USTE(Q,5)APEFs 计算的结果精度更高。最后,通过CBS/USTE(Q,5)和CBS/USTE(Q,5)APEFs 求解径向薛定谔方程,首次计算出SiH(X2П)(J=0)的振动态,画出了振动能级图。综上所述,本文的结果提供了SiH(X2П)更准确的光谱参数和更完备的振动态信息。
