TFE3-expressing malignant perivascular epithelioid cell tumor of the mesentery:A case report and review of literature
2020-04-08NahIhmKimJiShinLeeYooDukChoiChulJuJongHeeNam
Nah Ihm Kim,Ji Shin Lee,Yoo Duk Choi,U Chul Ju,Jong Hee Nam
Nah Ihm Kim,Yoo Duk Choi,Jong Hee Nam,Department of Pathology,Chonnam National University Hospital and Medical School,Gwangju 61469,South Korea
Ji Shin Lee,Department of Pathology,Chonnam National University Hwasun Hospital and Medical School,Hwasun 58128,South Korea
U Chul Ju,Department of Obstetrics and Gynecology,Chonnam National University Hwasun Hospital and Medical School,Hwasun 58128,South Korea
Abstract BACKGROUND Perivascular epithelioid cell tumor(PEComa)is a rare mesenchymal tumor that exhibits an epithelioid and spindle cell morphology.The tumor is characterized by immunoreactivity for melanocytic and myogenic markers but can be misdiagnosed as more common tumors with similar characteristics,including gastrointestinal stroma tumors or leiomyosarcomas.Recently,a subset of PEComas has been reported to harbor a transcription factor binding to TFE3 fusion.Herein,we report a rare case of TFE3-expressing malignant PEComa arising from the mesentery.CASE SUMMARY A 50-year-old woman presented with abdominal discomfort for 3 months.Results of laboratory tests were all within the normal ranges,and the patient had no notable medical history.Magnetic resonance imaging revealed a large tumor on the right side of the pelvic floor,which was originally suspected to be a primary ovarian tumor.However,during surgery,the tumor was revealed to have originated from the mesentery.Histologically,the tumor was composed of bundles of spindle cells and sheets of epithelioid cells.Extensive coagulative necrosis and numerous mitotic figures were observed.Immunohistochemistry revealed that the tumor cells were positive for smooth muscle actin,HMB-45,and TFE3 expression.Tumor involvement of the rectal serosa was identified,leading to a final diagnosis of malignant PEComa of the mesentery.Surgical resection was followed by adjuvant chemotherapy.No recurrence or metastasis was observed over a 6-month follow-up period.CONCLUSION Malignant PEComa of the mesentery is extremely rare and should be distinguished from morphological mimics through differential diagnosis and immunohistochemistry.
Key Words:Perivascular epithelioid cell tumor;TFE3;Differential diagnosis;Mesentery;Histology;Case report
INTRODUCTION
Perivascular epithelioid cell tumor(PEComa)is an uncommon mesenchymal neoplasm.Bonettiet al[1,2]first described this rare tumor type in 1992,and Zamboniet al[3]subsequently coined the term “PEComa” in 1996.The World Health Organization recognized PEComa as a distinct tumor entity in 2002[4].In 2005,Folpeet al[5]first reported four patients with PEComa of the mesentery.Including this first case series,to the best of our knowledge,only nine cases of primary PEComa of the mesentery have been reported to date[5-10].Since PEComa is a rare disease,other more common tumors such as gastrointestinal stroma tumor or leiomyosarcoma that show similar morphological characteristics will first be considered in the context of a differential diagnosis.
Histologically,PEComas are composed of epithelioid and spindle cells with radial growth around blood vessels.The characteristic expression of myogenic and melanocytic markers supports a diagnosis of PEComa[11].Recently,molecular studies identified a subset of PEComas with transcription factor binding toTFE3rearrangement,suggesting that PEComas harboringTFE3fusions may represent a distinct disease entity[12].
Herein,we describe a rare case of malignant PEComa originating from the mesentery and provide guidance for improving differential diagnosis of this tumor along with a review of the relevant literature.
CASE PRESENTATION
Chief complaints
A 50-year-old woman was admitted to the gynecology department of Chonnam National University Hospital with complaints of abdominal discomfort.
History and present illness
The patient exhibited symptoms 3 months prior to admission,and initial sonography performed in a primary health care center revealed an ovarian tumor of 8 cm × 6 cm ×6 cm at that time.The patient was then referred to a tertiary hospital for further evaluation and treatment.
History of past illness
The patient had no history of diabetes,hypertension,or other gynecological disease.
Personal and family history
The patient had no significant personal or family medical history.
Physical examination
The patient did not exhibit weight loss,palpable lymphadenopathy,or organomegaly.
Laboratory examinations
Results of laboratory tests,including carcinoembryonic antigen and cancer antigen 125 levels,and Risk of Ovarian Malignancy Algorithm score were all in the normal range.
Imaging examinations
Magnetic resonance imaging revealed a large heterogeneous mass(8 cm × 6.5 cm × 7 cm)occupying the pelvic cavity(Figure 1).Based on this finding and location,the preoperative diagnosis was malignant ovarian tumor.A chest computed tomography(CT)scan showed no evidence of distant metastasis to the thorax.
FINAL DIAGNOSIS
The final diagnosis was malignant PEComa of the mesentery with rectal involvement.
TREATMENT
The patient underwent total hysterectomy with bilateral salpingo-oophorectomy and pelvic lymph node dissection.A large mass was identified on the right side of the pelvic cavity,which was attached to the mesentery.The right ovary and fallopian tube were grossly normal.The rectum was also partially resected owing to suspicion of tumor involvement in the serosa.
Upon macroscopic examination,the lesion appeared as a white-to-tan solid mass.A cross section of the tumor revealed that the mass was lobulated,did not have a fibrous capsule,and had a focal area of necrosis(Figure 2).
Histologically,the tumor appeared as a high-grade spindle cell sarcoma with involvement of the rectal serosa.No metastases were found in the regional lymph nodes.The tumor was composed of fascicles of spindle cells with an infiltrative growth pattern.The majority of the tumor cells exhibited marked atypia with dispersed chromatin and macronucleoli.Extensive coagulative necrosis was observed with substantial mitotic activity detected reaching up to 40 mitoses per 10 high-power fields.Epithelioid foci were also identified.Nests of large epithelioid cells were surrounded by a delicate vasculature in a radial manner.The tumor cells displayed large,vesicular nuclei with prominent nucleoli and an abundant eosinophilic granular cytoplasm with striking pleomorphism(Figure 3).
Immunohistochemistry revealed that the tumor expressed HMB45,smooth muscle actin,andTFE3,but was negative forMelan A,Desmin,CD117,CD34,CD68,Myogenin,S100,MDM2,andCDK4(Figure 3).Thus,histopathological and immunohistochemical examinations confirmed the diagnosis of malignant PEComa.
Subsequently,whole-body positron emission tomography/CT scan were performed to check for possible metastases;however,no abnormality was found.Following surgery,the patient was treated with sirolimus chemotherapy.
OUTCOME AND FOLLOW-UP
No recurrence or metastasis was observed during a 6-month follow-up period.
DISCUSSION
PEComa is a rare mesenchymal tumor composed of histologically and immunohistochemically distinct perivascular epithelioid cells.The tumor cells exhibit an epithelioid and spindle morphology and express melanocytic and myogenic markers[11].Although the histogenesis and the normal counterpart of PEComa remain unknown,the term “PEComa” is widely accepted to include angiomyolipoma of the kidney,clear-cell “sugar” tumor or lymphangioleiomyomatosis of the lung,and other tumors originating from various anatomical sites[13,14].
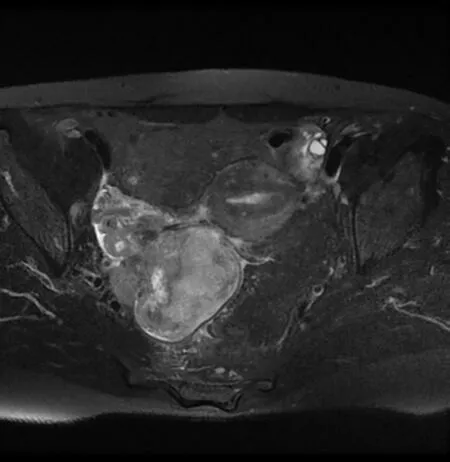
Figure 1 Pelvis magnetic resonance imaging showing heterogeneously enhancing mass(8 cm × 6 cm × 6 cm)occupying right pelvic cavity.

Figure 2 Gross findings exhibiting a relatively well-circumscribed white to tan solid mass with focal necrosis.
PEComas have been described in various organs;however,the majority are gynecological and gastrointestinal in origin[5,15-17].Mesenteric PEComas are extremely rare,with only nine cases reported to date.A literature review revealed that mesenteric PEComas affect women more often than men(male:female = 20:80),regardless of age[5-10].Seven out of ten reported cases,including the present case,were considered to be malignant PEComa exhibiting worrisome features,with only one case showing lymph node involvement.The majority of cases diagnosed as malignant PEComa were treated by surgical resection followed by adjuvant chemotherapy;however,the tumors recurred within 6-22 months in two cases[5-10](Table 1).
Microscopically,these tumors consist of bundles of spindle cells and sheets of epithelioid cells located around the blood vessels,and the tumor cells show striking pleomorphism with elevated mitotic activity.Such tumors with unusual locations and histopathological features may pose a diagnostic challenge,and differential diagnosescan include leiomyosarcoma,clear cell sarcoma of the soft tissue,alveolar soft part sarcoma(ASPS),malignant melanoma,gastrointestinal stromal tumor,and dedifferentiated liposarcoma.
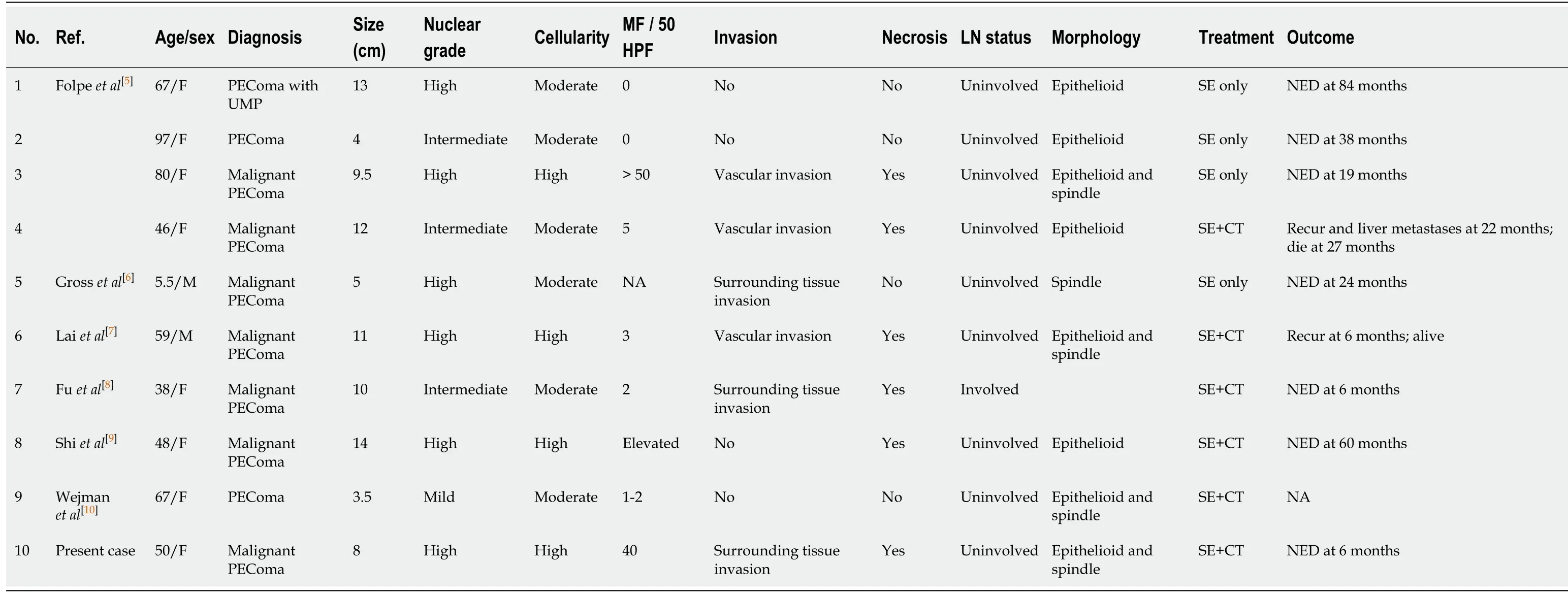
Table 1 Clinicopathologic features of mesenteric perivascular epithelioid cell tumor described previous and in present reports
Leiomyosarcomas should be considered according to the location and histological features of the tumor.PEComas and leiomyosarcomas are morphologically similar as they are both composed of spindle and/or epithelioid cells displaying varying degrees of atypia with positive reactivity for smooth muscle markers.However,leiomyosarcomas are consistently negative for melanocytic markers[18,19].
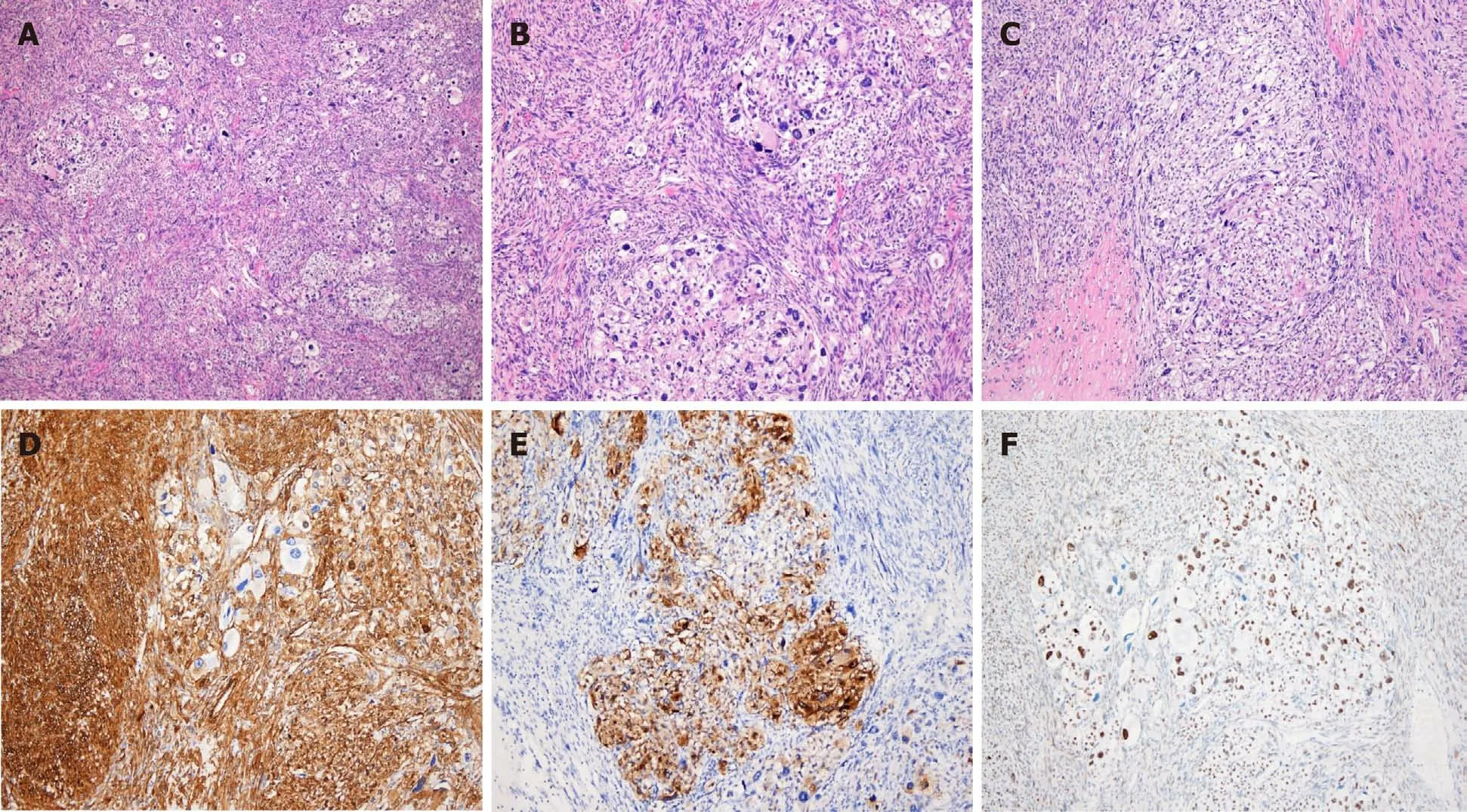
Figure 3 Histopathologic and immunohistochemical findings.A:The tumor was composed of sheets of epithelioid cells and bundles of spindle cells,hematoxylin and eosin(H&E),× 40;B and C:Mixed epithelioid and spindle cells exhibiting striking pleomorphism and necrosis(H&E,× 100);D:Spindle cells were positive for smooth muscle actin(× 100);E:Epithelioid cells were positive for HMB-45(× 100);F:Epithelioid cells showed nuclear positivity of TFE3(× 100).
Clear cell sarcoma of the soft tissue and malignant melanoma may also exhibit similar histological features to PEComa,along with positive immunohistochemical staining of melanocytic markers[20].However,clear cell sarcomas of the soft tissue display cellular nests separated by a fibrocollagenous network and strong S100 expression,whereas PEComas typically show a delicate vascular rich stroma and do not express S100.ASPS is a rare malignant soft tissue neoplasm with a strong predilection to develop in young adults and adolescents.The tumor shares both histological and immunohistochemical features with PEComa.ASPS is composed of large epithelioid cells with abundant cytoplasm,arranged in nests or sheets that are separated by a delicate vascular network of capillaries[21].However,ASPS can be differentiated from PEComa by immunohistochemical staining based on negative staining for melanocytic markers,which aids in differentiating from PEComa[20].The differential diagnosis of PEComa from gastrointestinal stromal tumors and dedifferentiated liposarcomas is based on the absence of CD117,DOG-1,MDM2,andCDK4expression.
The tumor in the present case showed positive immunoreactivity for melanocytic and myogenic markers and was negative for S100,Desmin,CD34,CD117,MDM2,and PAX-8.Further investigation ofTFE3immunoreactivity and its nuclear expression led to the ultimate diagnosis of malignant PEComa of the mesentery.
TFE3is a member of the microphthalmia-associated transcription factor family,and its expression is upregulated in ASPS and translocation-associated renal cell carcinomas[22].Since Folpeet al[5]first describedTFE3expression in PEComa,molecular analyses have identified a subgroup of PEComas withTFE3gene rearrangement,suggesting that PEComas harboringTFE3fusions may represent a distinct disease[12,23-28].
The majority of PEComas are composed of epithelioid and spindle cells,exhibiting positive myogenic expression and the presence of spindle cell components.In addition,PEComas are typically associated with tuberous sclerosis,whereas the subsect of PEComas withTFE3translocation have different histological features,exhibiting a predominantly epithelioid nested or alveolar architecture without spindle cell components,and immunohistochemistry reveals a lack of myogenic markers[29,30].Compared with the conventional type,TFE3-translocated PEComas typically have been reported in younger patients without a history of tuberous sclerosis complex[12,31-33].However,Williamsonet al[32]reported aTFE3-rearranged PEComa showing both an epithelioid and spindle morphology with expression of smooth muscle actin,which are consistent with the findings of the present case.
PEComa is a neoplasm of uncertain malignant potential.Although the majority of PEComas show a benign course,some are potentially malignant.However,there are no established criteria or markers to predict the clinical behavior of the tumor.Folpeet al[5],as well as Bleekeret al[34],proposed criteria for the classification of PEComas with worrisome features,including a tumor size > 5 cm,infiltrative growth pattern,high nuclear grade,cellularity,necrosis,vascular invasion,and a mitotic rate > 1/50 high-power fields.The tumor in the present study was 7 cm in size,had extensive coagulative necrosis,and showed a mitotic rate of 40/10 high-power fields.Therefore,the tumor met the aforementioned criteria for high risk of malignancy.Furthermore,the tumor involved the rectal serosa but no lymph node metastasis was observed.Therefore,a diagnosis of malignant PEComa was made based on the histopathological and immunohistochemical features.
Since PEComa is a very rare neoplasm,there are currently no effective therapies or management strategies.Surgical resection is the most common approach for curative treatment,and adjuvant chemotherapy is recommended for patients with tumors exhibiting malignant features.This unusual case of malignant PEComa of the mesentery further expands this field to help inform the diagnosis and treatment of this rare entity.
CONCLUSION
PEComas are rare mesenchymal neoplasms and should be thoroughly distinguished from their morphological mimics.Analysis of histopathological features and immunohistochemistry are essential for a differential diagnosis.The present case further emphasizes the importance of differential diagnosis based on the correct use of immunohistochemistry along with close surveillance of this unique tumor entity.Further studies are required to gain insight into the pathogenesis of PEComa and its clinical behavior.
ACKNOWLEDGEMENTS
We are grateful to the patient for allowing us to use her medical records in our case report.
杂志排行

World Journal of Clinical Cases的其它文章
- Special features of SARS-CoV-2 in daily practice
- Gastrointestinal insights during the COVID-19 epidemic
- From infections to autoimmunity:Diagnostic challenges in common variable immunodeficiency
- One disease,many faces-typical and atypical presentations of SARS-CoV-2 infection-related COVID-19 disease
- Application of artificial neural networks in detection and diagnosis of gastrointestinal and liver tumors
- Hepatic epithelioid hemangioendothelioma:Update on diagnosis and therapy