影响前列腺癌风险的关键基因识别
2020-04-07李熹阳谷明宇华琳
李熹阳 谷明宇 华琳

摘要:目的 基于TCGA数据库筛选影响前列腺癌(PCa)风险水平的关键基因,并建立PCa患者生存风险预测模型。方法 从TCGA数据库下载PCa患者基因表达数据及相关临床数据,通过前期研究初步筛选基因,并将患者分为高、低风险两类;对基因进行差异表达分析和GO和KEGG通路富集分析,筛选相关基因和信号通路;对差异表达基因进行蛋白互作网络分析,标记出关键基因;将关键基因的表达数据与PCa患者生存时间纳入Cox回归分析,建立生存风险预测模型。结果 前期研究得到620个基因,高风险患者234例,低风险患者285例;差异表达分析获得30个基因,主要分子功能(MF)为:受体结合和生长因子活动,生物学过程(BP)主要为细胞-细胞信号传导、细胞增殖的积极调节、血管生成的调节和细胞表面受体信号通路,细胞组分(CC)主要定位于细胞外區域,而KEGG信号通路为细胞因子-细胞因子受体相互作用;蛋白互作分析中共7个基因有相互作用,Cytoscape筛选出5个关键基因:PHYHIPL、CNTFR、GFRA1、EDN3和PROK1。结论 通过本研究识别的影响PCa预后的关键基因,发现潜在的PCa风险靶点,可能为PCa的治疗和预后提供帮助。
关键词:前列腺癌;基因差异表达分析;生物信息学;富集分析
中图分类号:R737.25 文献标识码:A DOI:10.3969/j.issn.1006-1959.2020.02.022
文章编号:1006-1959(2020)02-0080-06
Abstract:Objective To screen the key genes affecting prostate cancer (PCa) risk level based on the TCGA database and establish a survival risk prediction model for PCa patients. Methods Download PCa patient gene expression data and related clinical data from the TCGA database, preliminary screening of genes through early research, and classify patients into high and low risk categories; perform differential expression analysis of genes and enrichment analysis of GO and KEGG pathways to screen relevant gene and signal pathway; Perform protein interaction network analysis on differentially expressed genes to mark key genes; incorporate expression data of key genes and PCa patient survival time into Cox regression analysis to establish a survival risk prediction model. Results 620 genes were obtained in previous studies, 234 patients were high-risk patients, 285 patients were low-risk patients; 30 genes were obtained by differential expression analysis. The main molecular functions (MF) were: receptor binding and growth factor activity, and the main biological process (BP) For cell-cell signaling, positive regulation of cell proliferation, regulation of angiogenesis, and cell surface receptor signaling pathways, the cell component (CC) is mainly located in the extracellular region, while the KEGG signaling pathway is a cytokine-cytokine receptor interactions: A total of 7 genes interacted in the protein interaction analysis. Cytoscape screened out 5 key genes: PHYHIPL, CNTFR, GFRA1, EDN3, and PROK1.Conclusion The key genes affecting the prognosis of PCa identified through this study, and the discovery of potential PCa risk targets may help the treatment and prognosis of PCa.
Key words:Prostate cancer;Differential gene expression analysis;Bioinformatics;Enrichment analysis
前列腺癌(prostate cancer,PCa)是一种上皮性恶性肿瘤,是男性最为常见的癌症类型[1],国际癌症研究署(IARC)公开数据显示,2018年中国PCa标化发病率为13.9/10万,为低风险地区,但近10年来,PCa已成为我国增速最快的男性恶性肿瘤[2],并且由于人口老龄化进程加快,我国PCa发病率也高居男性恶性肿瘤第6位[3]。目前在PCa临床筛查与诊断中,广泛应用的血清前列腺特异抗原(PSA)检测敏感度高而特异性低[4],亟需寻找新的PCa标志物;癌症基因组图谱公共数据集(TCGA)数据库存有大规模的基因测序和患者相关临床指标,本研究基于TCGA数据库下载的前列腺癌组织中多种基因的mRNA三代测序数据,利用多种生物信息学和生物统计学方法,识别影响高、低风险前列腺癌形成的关键基因,以期建立了前列腺癌患者生存时间统计预测模型,以期为探索PCa分子治疗提供一定的参考。
1数据与方法
1.1数据库选择 选取在TCGA肿瘤数据库收录的前列腺癌患者基因表达level 3数据(截至2018年初)为研究对象。生物学信息注释数据库(Database for Annotation,Visualization,and Integrated Discovery,DAVID)6.8在线工具(https://david.ncifcrf.gov)用于对TCGA数据库中筛选出的差异表达基因进行基因本体论(GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)信号通路富集分析,其中GO分析包括细胞组分(CC),生物过程(BP)和分子功能(MF)三个部分。应用STRING(Search Tool for the Retrieval of Interacting Genes)数据库(https://string-db.org/cgi/input.pl),对差异表达基因编码蛋白构建相互作用网络,选择Cytoscape[5] 3.7.2中cytoHubba插件筛选蛋白互作网络中的关键基因。
1.2样本风险聚类 从TCGA数据库中下载得到20530个基因的表达数据,568例PCa患者及对应的生存时间与生存状态。基因表达数据已经通过Tophat2比对到染色体hg19上,并生成标准化的FPKM(fragments per kilobase of transcript per million fragments mapped)值。随后采用Cox风险比例回归模型提取与PCa患者生存有关的基因。剔除含有缺失数据的样本,并基于K均值聚类算法,将其分为两组。根据死亡事件发生的频率,分别命名两组样本为高风险组和低风险组[6]。
1.3数据分析 将前期研究获得的基因表达数据整理、录入R软件中,并绘制基因表达箱图,比较样本表达数据的分布情况。采用R软件limma[7]包,使用无监督聚类方法展示519例样本间的相似性,绘制多维标度分析(multidimensional scaling,MDS)图,并使用edgeR[8]包估算所有基因的离散度,即生物变异系数(biological coefficient of variation,BCV)的平方[9],以展示基因差异程度。利用limma包进行差异表达分析,对低风险PCa患者肿瘤组织和高风险PCa患者肿瘤组织中的差异表达基因进行筛选。筛选条件为:差异表达超过2倍(|log FC|>1),校正后P<0.1且P<0.05。针对上述条件筛选出的差异表达基因,分别使用ggplot2[10]软件包和gplots软件包绘制火山图与热图,验证并展示差异表达基因的结果。利用DAVID 6.8在线工具对差异表达基因进行GO富集分析和 KEGG信号通路富集分析,以P<0.05为标准筛选出具有显著性的差异表达基因功能注释和KEGG通路富集分析结果。在STRING数据库对上述筛选出的差异表达基因编码蛋白构建相互作用网络,条件为:①蛋白互作数据来源:Textmining,Experiments, Databases,Co-expression,Neighborhood,Gene Fusion,Co-occurrence;②最低作用评分要求为:0.4。将构建的蛋白互作网路数据导入Cytoscape中,利用cytoHubba插件查找hub节点,即相关信号通路中的关键基因。使用R软件rms包对筛选出的关键基因与PCa患者生存时間进行Cox回归分析,将关键基因的表达数据作为自变量,PCa患者生存时间作为因变量,并绘制诺莫图(Nomograph),建立统计预测模型,预测PCa患者的生存风险。
1.4统计学处理 使用R软件(3.6.1)进行统计学分析。通过绘制Kaplan-Meier曲线进行生存分析,并且使用对数秩检验法(Log-rank)检验显著性。差异表达分析采用经验贝叶斯先验趋势法(empirical bayes prior trend),亦即“limma-trend”法,对均值-方差关系建模。P<0.05表示差异有统计学意义。
2结果
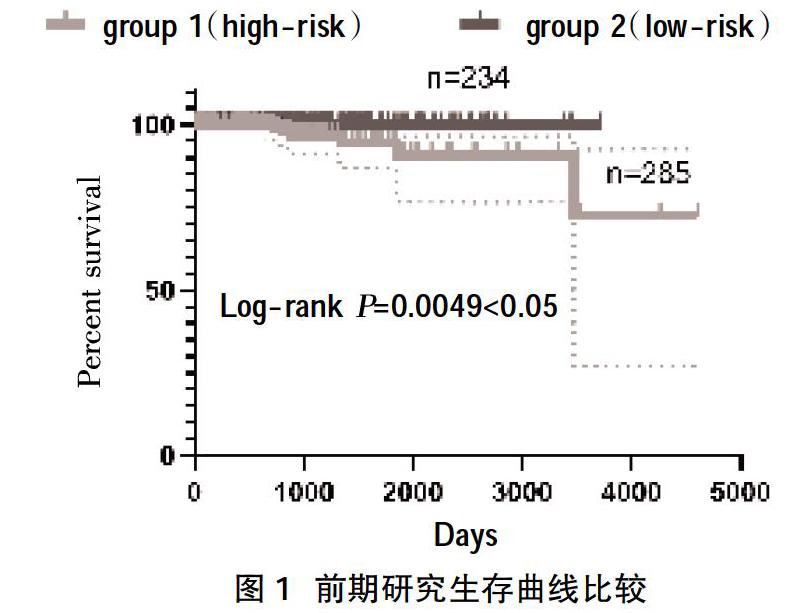
2.1样本风险聚类 经过单变量Cox分析,获得620个与前列腺癌患者生存相关的基因;K均值聚类算法将前列腺癌患者聚为两类,其中第一类(高风险)PCa患者234例,含7个死亡病例;第二类(低风险)PCa患者285例,没有死亡病例。绘制Kaplan-Meier曲线比较高风险(group1)与低风险(group2)PCa患者的生存率,采用Log-rank检验法测定生存率曲线间的显著性(图1),结果显示两条生存曲线之间存在统计学差异(P=0.004),说明通过聚类方法有效地将PCa患者聚为了高风险、低风险两类。
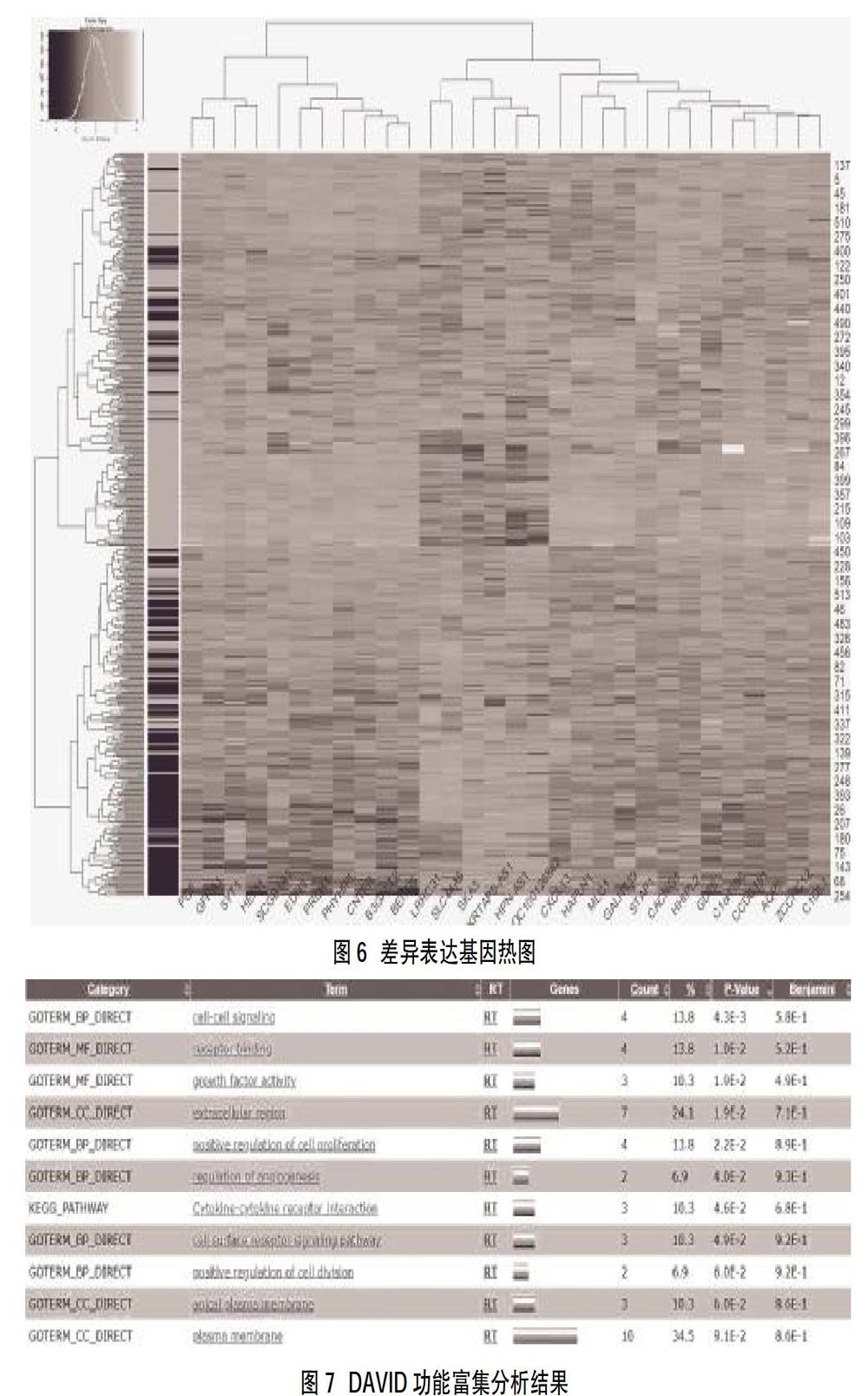
2.2差異表达基因筛选 基因表达箱式图,见图2,可见基因表达数据较为整齐,可以进行Limma分析; MDS图(图3)显示在前两个维度构成的坐标系中,所有样本相似度良好;所有基因的估计离散值(图4),包括经验贝叶斯稳健离散值(Tagwise)、经验贝叶斯稳健离散值的均值(Common)和经验贝叶斯稳健离散值的拟合值(Trended),曲线显示样本之间的差异较小。Limma筛选出30个差异表达基因,其中上调基因6个,下调基因24个(表1)。绘制上述差异表达基因的火山图(图5)。使用gplots软件包绘制热图(图6),横坐标为样本编号,纵坐标为基因种类,并对样本和基因双向聚类,展示表达差异分析结果。从图6左侧系统聚类树状图可见,30个表达差异基因恰好将519个样本分为两类;上调的基因显示为黄色,下调的基因显示为红色,6个上调基因:KRTAP5-AS1,LOC100128842,SKA3,HPN-AS1,SLC44A5和LRRC31表达情况明显异于其他基因,与差异表达分析得出的结果相符。
2.3差异表达基因的功能富集分析 以P<0.05筛选功能富集分析结果(图7):差异表达基因 MF主要为受体结合(P=0.010),生长因子活动(P=0.019);BP主要为细胞-细胞信号传导(P=0.0043),细胞增殖的积极调节(P=0.022),血管生成的调节(P=0.040),细胞表面受体信号通路(P=0.049);CC主要为细胞外区域(P=0.019),KEGG信号通路为细胞因子-细胞因子受体相互作用(P=0.046)。
腺和胎盘)中表达,该基因编码的蛋白质诱导腺体中的毛细血管内皮细胞的增殖、迁移。Jilg CA等[19]研究发现,PRK1(即PROK1)基因有望成为治疗雄激素非依赖性转移性PCa的治疗靶标。
本研究还基于上述关键基因表达与生存时间的Cox回归构建了统计预测模型。诺莫图是一个复杂数学公式的图形表示[20],在肿瘤学和医学中被广泛被用作预测手段,是现代医学决策制定的重要组成部分,可以通过综合不同预后和决定性变量,产生个体发生临床事件的数字概率[21]。本研究绘制出的诺莫图具有良好的区分度,C-index达到了0.729,不同关键基因表达水平组合的患者可以得到良好的区分。
参考文献:
[1]Howard N,Clementino M,Kim D,et al.New developments in mechanisms of prostate cancer progression[J].Semin Cancer Biol,2019(57):111-116.
[2]前列腺癌成中国增速最快的男性恶性肿瘤[J].肿瘤防治研究,2019,46(7):666.
[3]王宁,刘硕,杨雷,等.2018全球癌症统计报告解读[J].肿瘤综合治疗电子杂志,2019,5(1):87-97.
[4]王炜,李传刚,刘辉,等.前列腺特异性抗原对前列腺癌诊断价值的探讨[J].中国医科大学学报,2016,45(1):61-65,69.
[5]Shannon,P.Cytoscape:A Software Environment for Integrated Models of Biomolecular Interaction Networks[J].Genome Research,2003,13(11):2498-2504.
[6]Hua L,Xia H,Xu W,et al.Risk stratification for prostate cancer via the integration of omics data of The Cancer Genome Atlas[J].Translational Cancer Research,2018,7(3):706-719.
[7]Phipson B,Lee S,Majewski IJ,et al.Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression[J].The Annals of Applied Statistics,2016,10(2):946-963.
[8]Robinson MD,Smyth GK.Moderated statistical tests for assessing differences in tagabundance[J].Bioinformatics,2008,23(21):2881-2887.
[9]Mccarthy DJ,Chen Y,Smyth GK.Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation[J].Nucleic Acids Research,2012,40(10):4288-4297.
[10]Wickham H.ggplot2:elegant graphics for data analysis[J].J R Stat Soc,2011,174(1):245-246.
[11]Hendriks RJ,Dijkstra S,Smit FP,et al.Epigenetic markers in circulating cell-free DNA as prognostic markers for survival of castration-resistant prostate cancer patients[J]. Prostate,2018,78(5):336-342.
[12]Yin X,Yu J,Zhou Y,et al.Identification of CDK2 as a novel target in treatment of prostate cancer[J].Future Oncology,2018,14(8):709-718.
[13]蔣宏毅,赵晓昆,钟朝晖,等.PTEN/MMAC1/TEP1、TGF-β1在前列腺癌及前列腺增生中的表达及其意义[J].中国现代医学杂志,2008,18(9):1221-1225.
[14]Hearn JWD,Abuali G,Magi-Galluzzi C,et al.HSD3B1 and resistance to androgen deprivation therapy in prostate cancer[J].Journal of Clinical Oncology,2015,33(7_suppl):156-156.
[15]Fu H,Ge B,Chen D,et al.Phytanoyl-CoA 2-Hydroxylase-Interacting Protein-Like Gene Is a Therapeutic Target Gene for Glioblastoma Multiforme[J].Med Sci Monit,2019(25):2583-2590.
[16]Cousinery Mary C,LiRuili,VannitambyAmanda,et al.Neurotrophin signaling in a genitofemoral nerve target organ during testicular descent in wmice[J].Journal of Pediatric Surgery,2016,51(8).
[17]Grasso M,Fuso A,Dovere L,et al.Distribution of GFRA1-expressing spermatogonia in adult mouse testis[J].Reproduction,2012,143(3):325-332.
[18]谢萍芳,赵东怡,周美容,等.乳腺癌中GFRA1表达临床意义的生物信息学分析[J].中国肿瘤临床,2018,45(15):769-773.
[19]Jilg CA,Ketscher A,Metzger E,et al.PRK1/PKN1 controls migration and metastasis of androgen-independent prostate cancer cells[J].Oncotarget,2014,5(24):12646-12664.
[20]Grimes David A.The nomogram epidemic:resurgence of a medical relic[J].Annals of Internal Medicine,2008,149(4):273-275.
[21]Vinod P Balachandran,MithatGonen,J Joshua Smith,et al. Nomograms in oncology:more than meets the eye[J].The Lancet Oncology,2015,16(4):e173-e175.
收稿日期:2019-10-23;修回日期:2019-11-10
编辑/肖婷婷
