Discontinuous polyostotic fibrous dysplasia with multiple systemic disorders and unique genetic mutations: A case report
2020-04-07TiaoLinXinYuLiChangYeZouWeiWeiLiuJunFanLinXinXinZhangSiQiZhaoXianBiaoXieGangHuangJunQiangYinJingNanShen
Tiao Lin, Xin-Yu Li, Chang-Ye Zou, Wei-Wei Liu, Jun-Fan Lin, Xin-Xin Zhang, Si-Qi Zhao, Xian-Biao Xie, Gang Huang, Jun-Qiang Yin, Jing-Nan Shen
Tiao Lin, Chang-Ye Zou, Xian-Biao Xie, Gang Huang, Jun-Qiang Yin, Jing-Nan Shen, Department of Musculoskeletal Oncology, The First Affiliated Hospital of Sun Yat-sen University,Guangzhou 510080, Guangdong Province, China
Xin-Yu Li, Wei-Wei Liu, Jun-Fan Lin, Xin-Xin Zhang, Si-Qi Zhao, Zhongshan School of Medicine,Sun Yat-sen University, Guangzhou 510080, Guangdong Province, China
Abstract BACKGROUND Polyostotic fibrous dysplasia (PFD) is an uncommon developmental bone disease in which normal bone and marrow are replaced by pseudotumoral tissue. The etiology of PFD is unclear, but it is generally thought to be caused by sporadic,post-zygotic mutations in the GNAS gene. Herein, we report the case of a young female with bone pain and lesions consistent with PFD, unique physical findings,and gene mutations.CASE SUMMARY A 27-year-old female presented with unbearable bone pain in her left foot for 4 years. Multiple bone lesions were detected by radiographic examinations, and a diagnosis of PFD was made after a biopsy of her left calcaneus with symptoms including pre-axial polydactyly on her left hand and severe ophthalmological problems such as high myopia, vitreous opacity, and choroidal atrophy. Her serum cortisol level was high, consistent with Cushing syndrome. Due to consanguineous marriage of her grandparents, boosted whole exome screening was performed to identify gene mutations. The results revealed mutations in HSPG2 and RIMS1, which may be contributing factors to her unique findings.CONCLUSION The unique findings in this patient with PFD may be related to mutations in the HSPG2 and RIMS1 genes.
Key Words: Polyostotic fibrous dysplasia; Genetic mutation; Hypercortisolism; Drug resistance; Ophthalmological problems; Case report
INTRODUCTION
Fibrous dysplasia (FD) is a genetic neoplastic bone disorder where normal bone and marrow are replaced by osteofibrous connective tissue, leading to bone pain,deformity, and fractures[1]. While the condition is benign, surgery is necessary to alleviate pain and repair or stabilize the affected bones. FD accounts for approximately 7% of all benign tumor-like bone lesions[2]. There are three defined subtypes of FD: (1)Monostotic; (2) Polyostotic fibrous dysplasia (PFD); and (3) McCune Albright syndrome (MAS). Among these, monostotic is the most common (70%), while PFD and MAS are relatively rare[3].
Bisphosphonates are the primary medical treatment for FD. Although a 1994 study showed that intravenous pamidronate (60 mg per day over 3 d, every 6 mo) can result in the refilling of bone lesions and cortical thickening of some patients with FD, its effectiveness in controlling disease progression remains uncertain[4]. Drugs such as denosumab have been reported to be effective in reducing bone pain and slowing the lesion growth rate in patients with receptor activator of nuclear factor kappa-B ligand expression, however, its use is debatable when the side effects are considered in light of the therapeutic effects[1]. Other treatments include surgery, physical rehabilitation,and long-term conservative management and close monitoring[5]. As such, to develop better treatments for FD, a better understanding of the genetic and molecular mechanisms of FD is needed.
FD is generally thought to be caused by sporadic, post-zygotic mutations in theGNASgene, located on chromosome 20q13.3, which result in the activation of the signaling transduction pathway that generates cyclic adenosine monophosphate[6].
Herein, we report the case of a 27-year-old Chinese woman diagnosed as PFD with multiple discontinuous lesions, Cushing syndrome, and ophthalmological disorders.She was unresponsive to bisphosphonate treatment, and gene sequencing revealed mutations in theHSPG2andRIMS1genes.
CASE PRESENTATION
Chief complaints
The patient complained of increasing pain in her left foot and difficulty walking over the past 4 years.
History of present illness
Over a 4-year period, she experienced increasing pain in her left foot and difficulty walking. Radiographic examinations at local hospitals suggested a diagnosis of PFD,which was confirmed by a biopsy of her left calcaneus.
History of past illness
The patient had a history of frequent bone fractures since birth. She also presented with typical symptoms of Cushing syndrome and ophthalmological disorders including high myopia, vitreous opacity, and choroid atrophy. Hypothalamic amenorrhea and irregular menstruation were also present.
Personal and family history
The patient’s maternal grandparents were consanguineous (cousins).
Physical examination
The patient experienced pain on palpation of her left foot. No obvious swelling was observed, and the foot was neurovascularly intact. Signs of Cushing syndrome included abdominal obesity with thin arms and legs, acne, a round face, and a fat lump between the shoulders (Figure 1A-C)[7]. Her teeth were noted to be poorly developed (Figure 1D), and she had thumb duplication on her left hand (Figure 1E and F).
Laboratory examinations
Her serum calcium and phosphate levels were normal as well as the levels of hormones that regulate calcium metabolism, including parathyroid hormone, 25-hydroxy vitamin D, and osteocalcin. Her serum cortisol level was 1492.00 nmol/L,which was extremely high (reference range of 147.30-609.30 nmol/L), and her triglyceride and uric acid levels were also elevated (Table 1).
Imaging examinations
Skeletal scintigraphy showed multiple bone lesions on her left ischium, distal fibula,calcaneus, and talus (Figure 2A and B). Computed tomography (CT) revealed wellcircumscribed bone lucencies and ground-glass opacities in the calcaneus and talus(Figure 2C and D). No evidence of pituitary or adrenal lesions was identified on brain computed tomography.
FINAL DIAGNOSIS
Based on the patient’s clinical symptoms, imaging studies, and biopsy results(Figure 3), a diagnosis of PFD was made after consultations with musculoskeletal oncologists, radiologists, and pathologists. Her other symptoms and signs were considered likely to be caused by her unique genetic mutations.
TREATMENT
In 2016, the patient began intravenous zoledronic acid every 6 mo and received four doses, followed by sodium pamidronate every 3 mo and received three doses. She was then continued on oral alendronate weekly.
OUTCOME AND FOLLOW-UP
After evaluating the risk and expenses of surgery, the patient chose to continue bisphosphonate treatment with regular monitoring of disease progression. Over a 2-year period, the size of the lesions did not become markedly larger. However, dualenergy X-ray absorptiometry indicated a new lower bone mass in the proximal femur and the distal fibula as compared to an examination 2 years prior, suggesting possible disease progression. The treatments, however, did not alleviate the pain in her left foot, indicating resistance to the anti-bone resorption treatments. Nonsteroidal antiinflammatory drugs were prescribed for her left foot bone pain.
DISCUSSION
Herein, we presented the case of a young woman with PFD combined with multiplesystemic disorders and resistance to bisphosphonate treatment. PFD was confirmed by biopsy of her left calcaneus. However, her additional symptoms were markedly different from most patients with PFD.
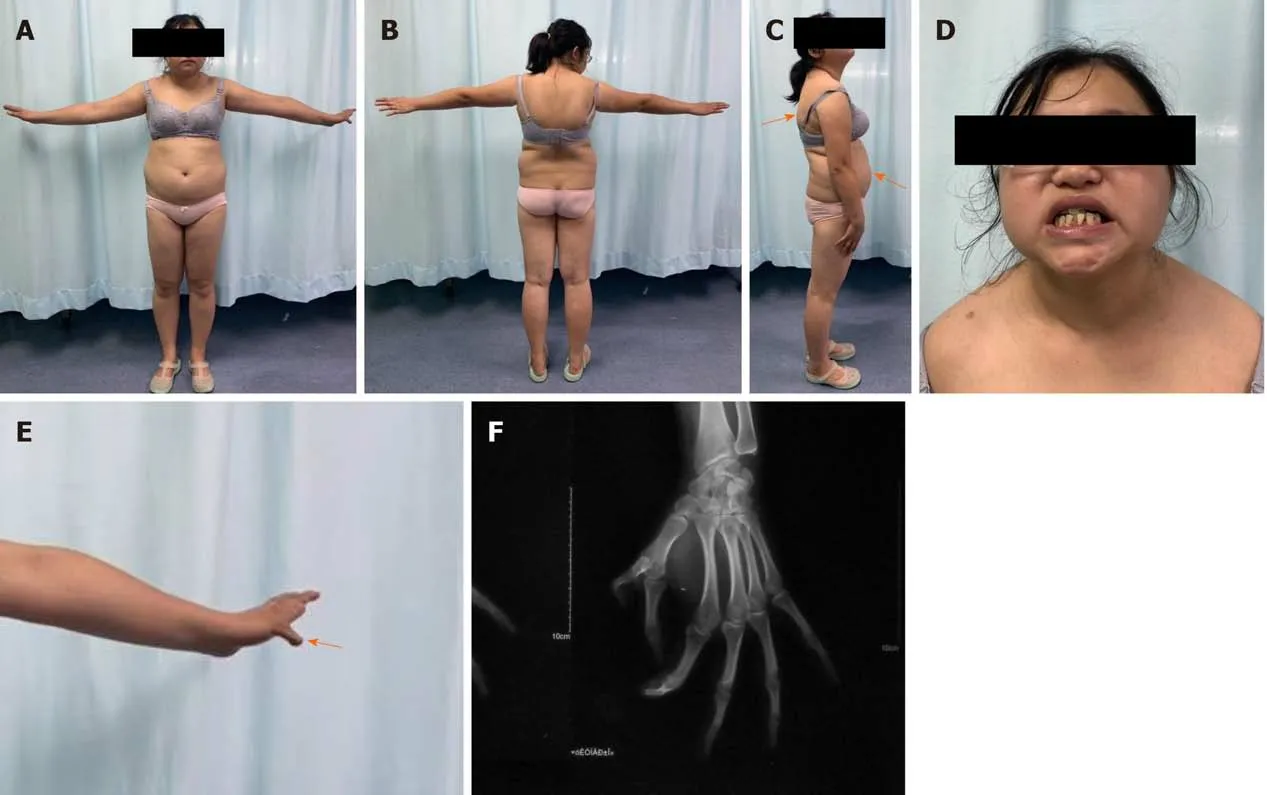
Figure 1 Photographic images of the patient showing signs of Cushing syndrome. A-C: Abdominal obesity with thin arms and legs, acne, a round face and a fat lump between the shoulders (orange arrows); D: Poorly developed teeth; E and F: Thumb duplication on the left hand (orange arrow).
We reviewed 6 cases of PFD identified in our search of the literature[2,8-12], and the details of the 6 cases along with the details of our case are summarized in Table 2. In patients with PFD, when lesions involve the orbital region the primary findings are facial asymmetry, orbital dystopia, and orbital proptosis[13], none of which were identified in our patient. Therefore, it is likely that the ophthalmological disorders of our patient are not related to her PFD, but rather to other genetic defects.
Some of the patients with PFD/MAS in our review did have multiple bone lesionsand endocrine disorders (e.g., precocious puberty, hypercortisolism, and hyperthyroidism) and skin pigmentation (“café-au-lait” spots). Hypercortisolism was the rarest symptom associated with PFD/MAS and is reported to occur exclusively in newborns[14]. However, our patient had an extremely high serum cortisol level, a finding not reported in the cases we reviewed.
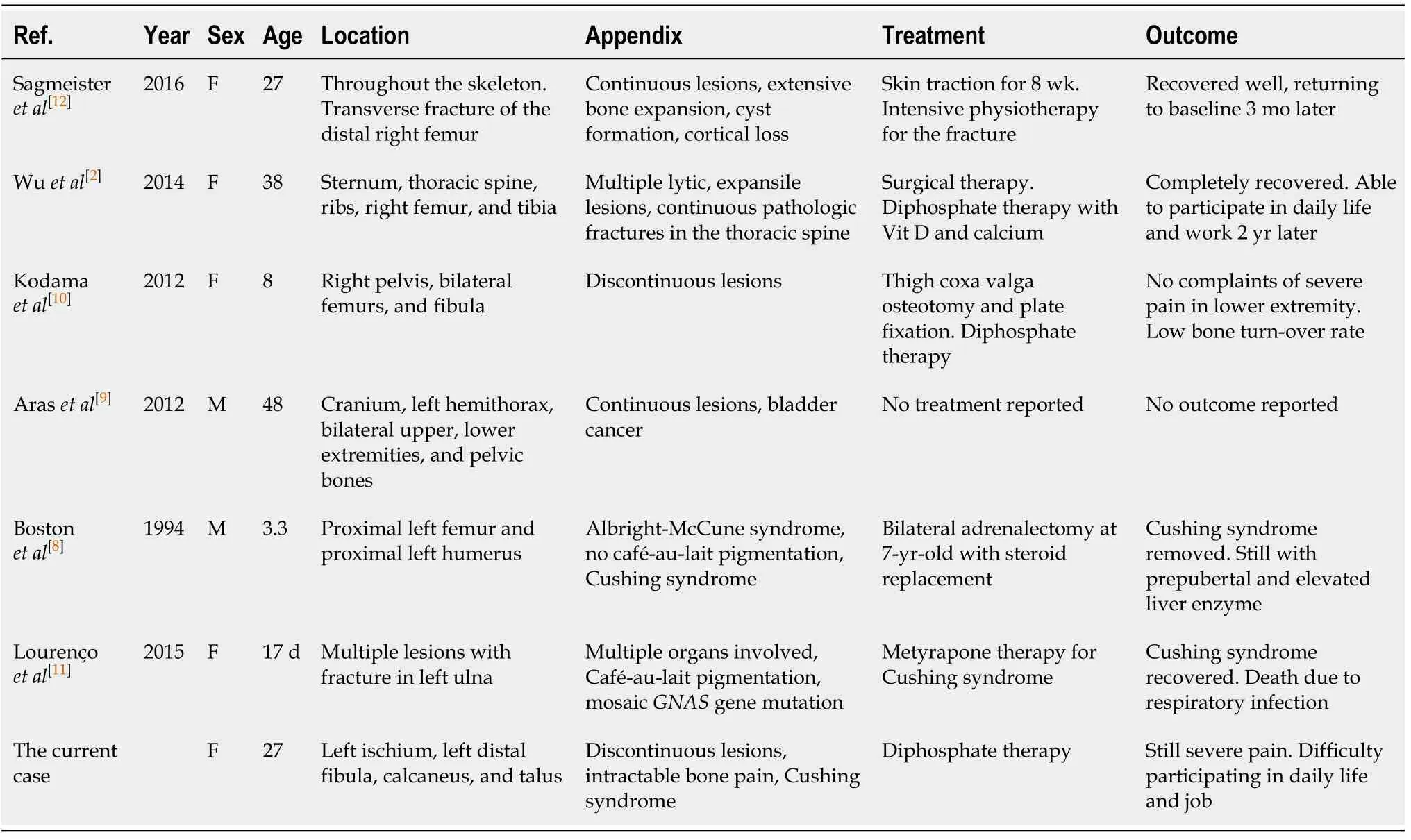
Table 2 Cases diagnosed as polyostotic fibrous dysplasia
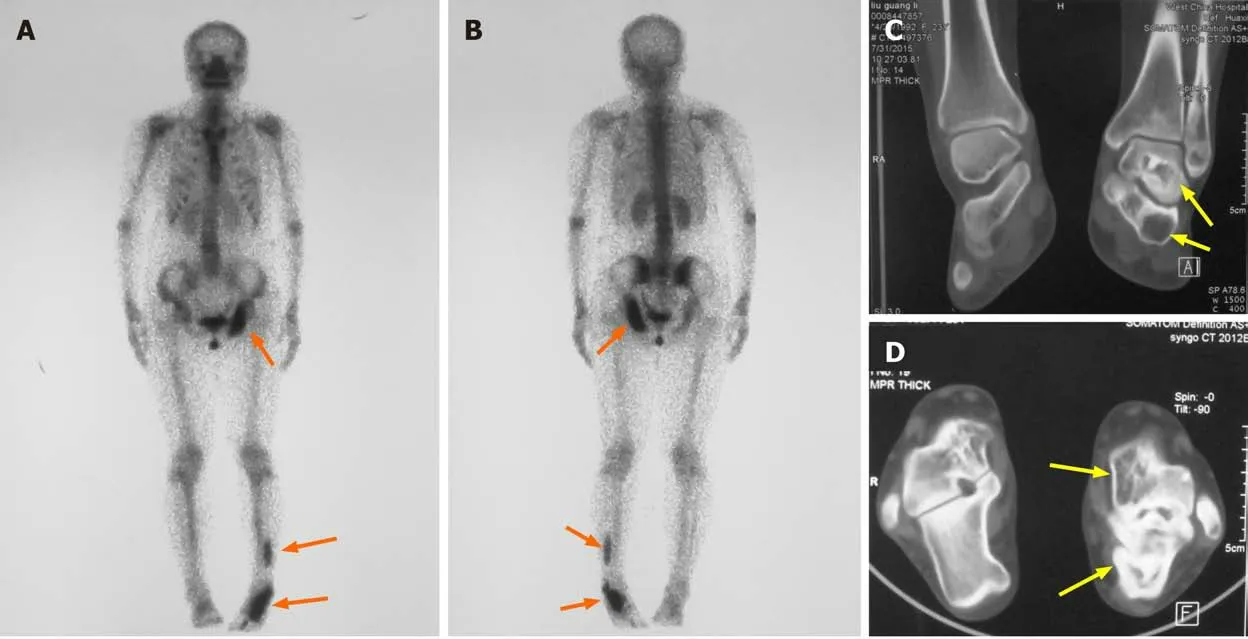
Figure 2 Radiography examination results. A: Anterior view; B: Posterior view of skeletal scintigraphy showing the location of the bone lesions on left ischium and left fibula, talus, and calcaneus (orange arrows); C and D: Computed tomography shows well-circumscribed bone lucencies and ground-glass opacities in left talus and calcaneus (yellow arrows).
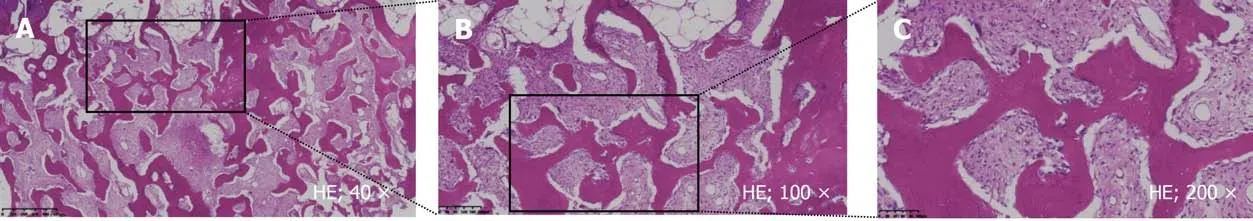
Figure 3 Pathological reports. A: Fibrous and osseous tissue are present in varying proportions [hematoxylin-eosin (HE), 40 ×]. The box shows the view field of B; B: The fibrous tissue is composed principally of bland fibroblastic cells. Mitoses are uncommon (HE, 100 ×). The box shows the view field of C; C: The osseous component is comprised of irregular, curvilinear, trabeculae of woven or rarely lamellar woven bone (HE; 200 ×).
PFD is difficult to treat or cure because of multiple advanced bone lesions and genetic defects of the osteoprogenitors. Studies have shown that radiographic findings and bone pain are improved in approximately 50% patients treated with bisphosphonates[15-17]. However, neither oral nor intravenous bisphosphonates were effective in our patient. Curettage is indicated to alleviate bone pain and improve limb function; however, our patient declined any surgical treatment.
Since multiple gene mutations were identified by boosted whole exome screening in our patient, we hypothesize that her unique symptoms are likely due to the gene mutations (Table 3). Notably, the missense and non-sense point mutations inRIMS1andHSPG2are likely responsible for her unique symptoms (Figure 4), because mutations in these two genes are reported to be associated with bone and ocular diseases, respectively.
Analysis of the genetic sequencing results of the patient and her parents indicated that the patient’s mutations were mostly heterozygous and similar to those of her mother. This indicates that the mutated genes came from her mother, whose parents had a consanguineous marriage. Surprisingly, theHSPG2mutation was homozygous,which is likely due to uniparental disomy[18][a single chromosome from her mother was duplicated leading to the homozygous mutation inHSPG2(Figure 5)].
HSPG2is an essential, highly conserved gene widely expressed throughout the development of cartilage and the formation and calcification of skeletal bone[19].Mutations inHSPG2can lead to two autosomal recessive inheritance skeletal disorders; Schwartz-Jampel syndrome (Online Mendelian Inheritance in Man 255800)and dys-segmental dysplasia, Silverman-Handmaker type (Online Mendelian Inheritance in Man 224410)[20]. Dys-segmental dysplasia, Silverman-Handmaker type can result in severe cartilage matrix anomalies, and even neonatal death, whereas patients with Schwartz-Jampel syndrome may have chondrodysplasias, myotonic myopathy, and facial and ocular abnormalities. Heterozygous mutations inRIMS1mainly cause cone-rod dystrophy, which is characterized by reduced photophobia,central vision, and reduced color vision[21].
We did not identify mutations inGNASin any of the blood samples we tested,although mutations inGNAShave been reported to be associated with PFD. It is generally thought that mutations inGNASoccur early in embryogenesis, and cells with defects are distributed in different tissues of the body as a result of embryonic cell migration[22]. To determine ifGNASmutations are present in our patient, bone lesion tissue samples require further sequencing analysis or molecular testing.
CONCLUSION
Herein, we presented the case of a patient with PFD with the unique findings of preaxial polydactyly, Cushing syndrome, ophthalmological abnormalities, and resistance to bisphosphonate treatment. Gene sequencing revealed mutations inHSPG2andRIMS1, which may be responsible for her unique findings.
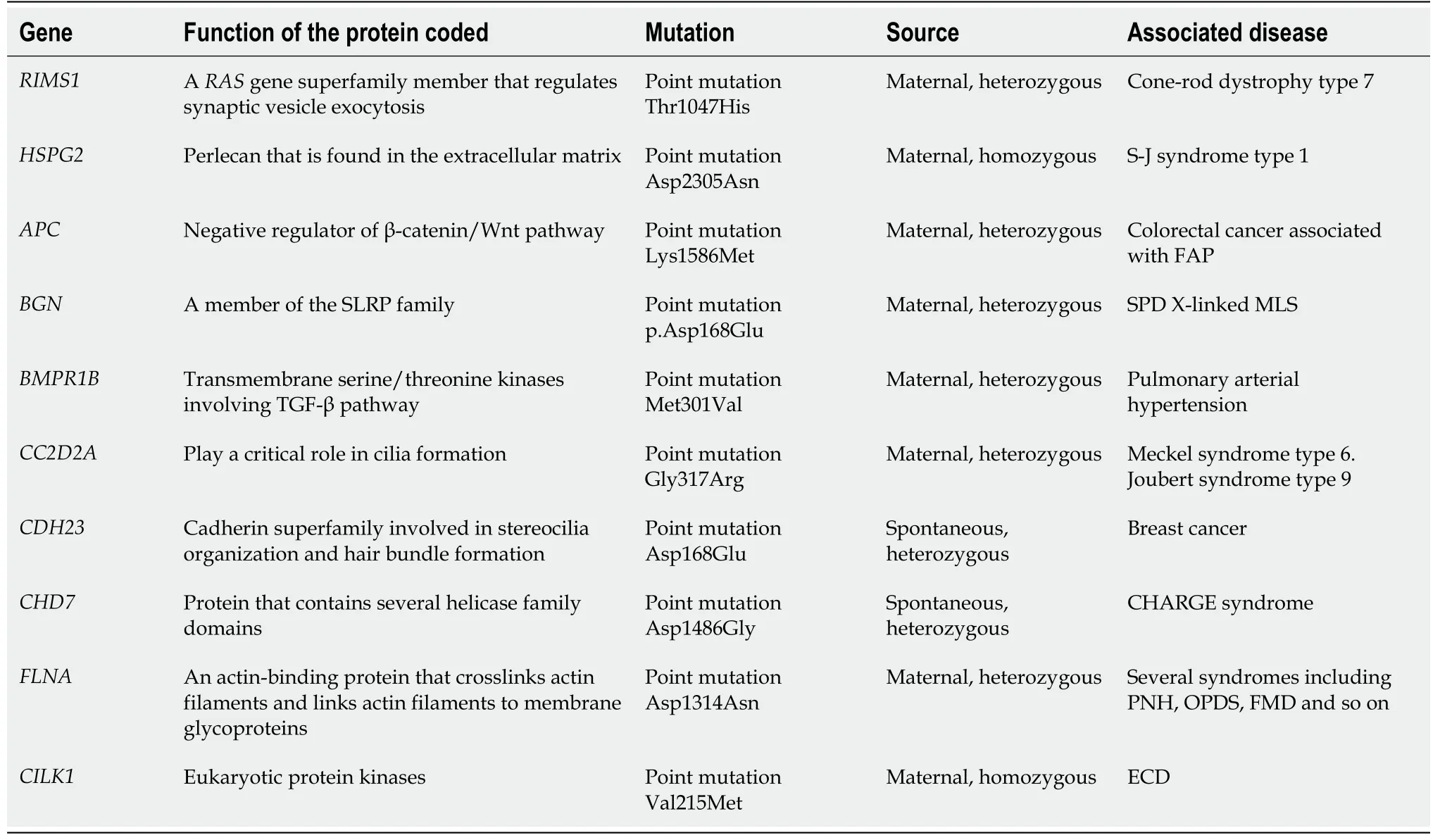
Table 3 Result of the boosted whole exome screening
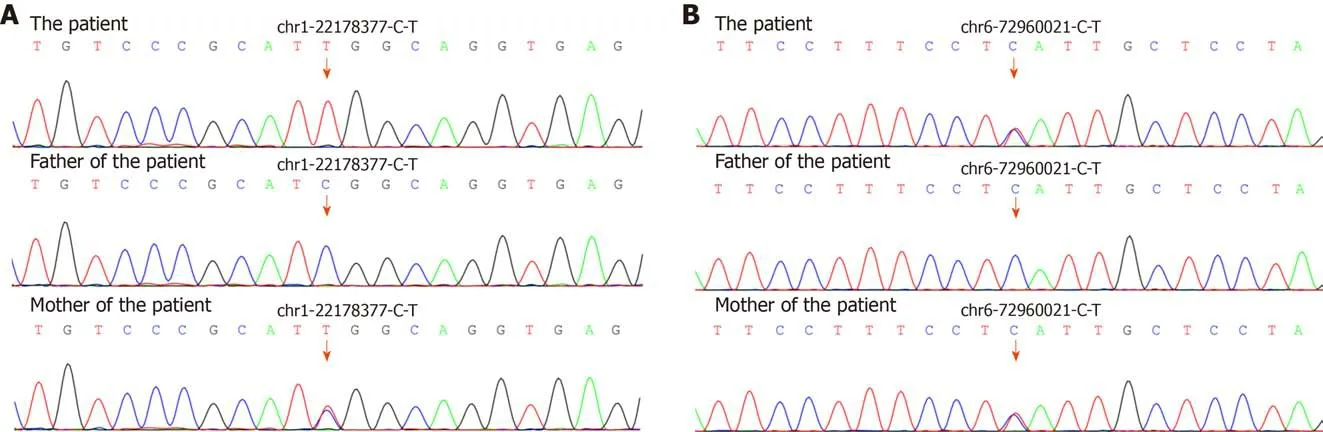
Figure 4 Sequencing chromatograms of the analyzed genes. A: Results for HSPG2; B: Results for RIMS1. The red arrows indicate the variants in HSPG2(c.6913G>A, p.Asp2305Asn) and RIMS1 (c.3139del, p.Thr1047His). The variants indicate a maternal source.
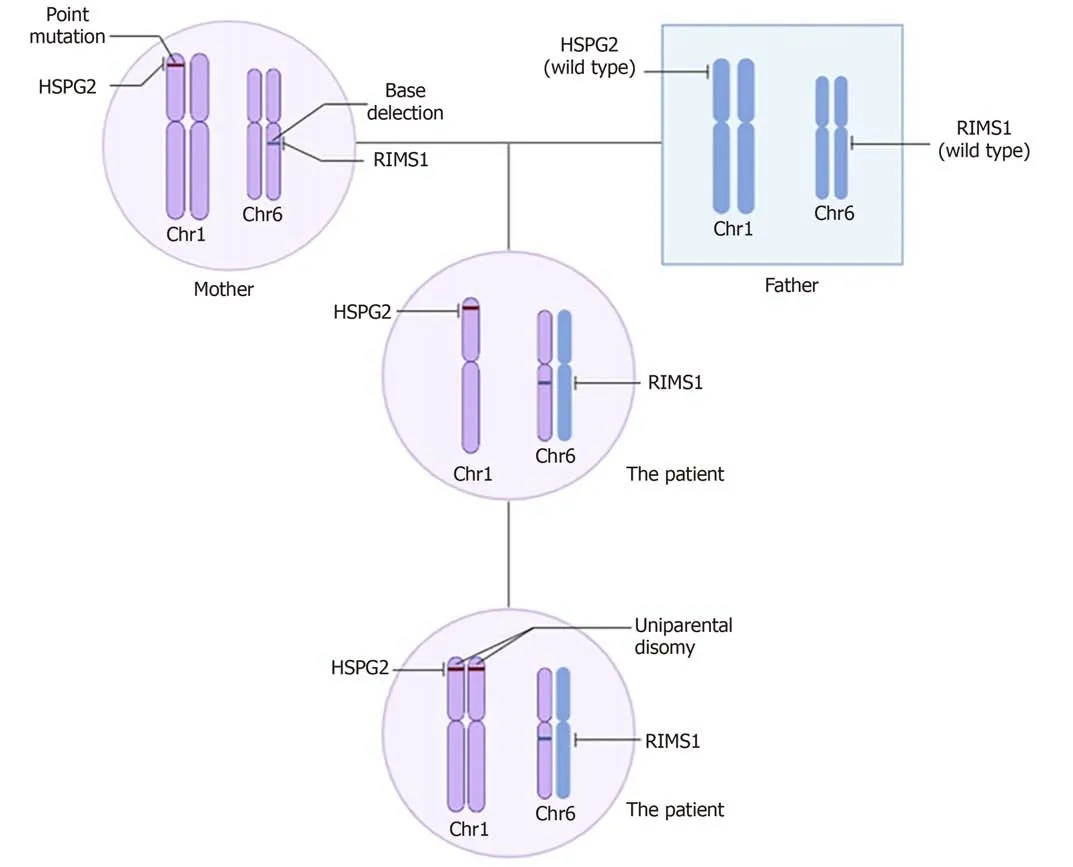
Figure 5 Possible genetic pedigree: The inheritance of HSPG2 and RIMS1. The HSPG2 mutation is homozygous, while the RIMS1 mutation is heterozygous. The abnormal genotype of HSPG2 is likely caused by uniparental disomy, a single chromosome from her mother is duplicated leading to the homozygous mutation in HSPG2.
杂志排行

World Journal of Clinical Cases的其它文章
- Novel triple therapy for hemorrhagic ascites caused by endometriosis: A case report
- Primary breast cancer patient with poliomyelitis: A case report
- Symptomatic and optimal supportive care of critical COVID-19: A case report and literature review
- Multiple ectopic goiter in the retroperitoneum, abdominal wall, liver,and diaphragm: A case report and review of literature
- Acute celiac artery occlusion secondary to blunt trauma: Two case reports
- Uncontrolled central hyperthermia by standard dose of bromocriptine: A case report