Submicroscopic 11p13 deletion including the elongator acetyltransferase complex subunit 4 gene in a girl with language failure, intellectual disability and congenital malformations: A case report
2020-04-07JaimeToralLopezLuzMarGonzlezHuertaOlgaMessinaBaasSergioCuevasCovarrubias
Jaime Toral-Lopez, Luz María González Huerta, Olga Messina-Baas, Sergio A Cuevas-Covarrubias
Jaime Toral-Lopez, Departamento de Genética Medica, Centro Medico Ecatepec, ISSEMYM,Ecatepec 55000, México
Jaime Toral-Lopez, Programa de Maestría y Doctorado en Ciencias Médicas, Odontológicas y de la Salud/Hospital Infantil de México, Universidad Nacional Autónoma de México, México 06720, México
Luz María González Huerta, Departamento de Biología Molecular, Hospital General de México,Cuauhtémoc 06720, México
Olga Messina-Baas, Departamento de Oftalmología, Hospital General de México, Cuauhtémoc 06720, México
Sergio A Cuevas-Covarrubias, Genetica, Hospital General de México, Cuauhtémoc 06726,Mexico
Sergio A Cuevas-Covarrubias, Programa de Maestría y Doctorado en Ciencias Médicas,Odontológicas y de la Salud, Universidad Nacional Autónoma de México, México 06720,Mexico
Abstract BACKGROUND We described the main features of an infant diagnosed with facial dysmorphic,language failure, intellectual disability and congenital malformations to strengthen our understanding of the disease. Currently, treatment is only rehabilitation and surgery for cleft lip and palate.CASE SUMMARY The proband was a 2-years-8-months-old girl. Familial history was negative for congenital malformations or intellectual disability. The patient had microcephaly,upward-slanting palpebral fissures, depressed nasal bridge, bulbous nose and bilateral cleft lip and palate. Brain magnetic resonance imaging showed cortical atrophy and band heterotopia. Her motor and intellectual development is delayed. A submicroscopic deletion in 11p13 involving the elongator acetyltransferase complex subunit 4 gene (ELP4) and a loss of heterozygosity in Xq25-q26.3 were detected.CONCLUSION There is no treatment for the ELP4 deletion caused by a submicroscopic 11p3 deletion. We describe a second case of deletion of the ELP4 gene without aniridia,which confirms the association between ELP4 gene with several defects and absence of this ocular defect. Additional clinical data in the deletion of the ELP4 gene as cleft palate, facial dysmorphism, and changes at level brain could be associated to this gene or be part of the effect of the recessives genes involved in the loss of heterozygosity region of Xq25-26.3.
Key Words: Submicroscopic 11p13 deletion; Elongator acetyltransferase complex subunit 4 gene; Language failure; Intellectual disability; Congenital malformations; Case report
INTRODUCTION
The elongator acetyltransferase complex subunit 4 gene (ELP4; MIM #606985) encodes the protein 4 of the elongator complex of ribonucleic acid polymerase II. ELP4 protein is composed of 424 amino acids and plays a role in transcriptional elongation, transfer ribonucleic acid modification, polarized exocytosis, and multiple types of cell migration. The failure of any member of the family of elongators, including ELP4, can be associated with different neurological disorders[1,2]. Twenty-five submicroscopic deletions, including theELP4gene and the adjacent sequences (excluding thePAX6gene, the neighboring gene related to aniridia), have been associated with intellectual disability, language development failure, autism spectrum disorder, and epilepsy with aniridia[3-9]or without aniridia[10]. TheELP4gene is 273.9 kb in size and encompasses 12 exons. Interestingly, inside the ultraconserved large intronic region between exons 9 to 12 of theELP4gene, there is a long-range cis-regulatory enhancer element located 25 to 150 kb downstream of thePAX6gene, which controls its expression[11,12]. In the present study, we described a girl with dysmorphia, language failure, intellectual disability,and congenital malformations without aniridia, and with a submicroscopic deletion in 11p13 affecting theELP4gene.
CASE PRESENTATION
Chief complaints
Registering a cleft lip and palate at 26 wk of gestation and delayed motor development at 2 years of age.
History of present illness
The patient, a 2-year-and-8-month-old Mexican girl, was brought by her parents for evaluation because of delays in her motor and language development and congenital malformations. Currently, her motor development is abnormal without head control,she still does not sit down. She also does not speak any words and often becomes ill from the respiratory tract without any serious complications.
History of past illness
The proband was the third child of two healthy, unrelated, and young parents (27 and 26 years old at the time of delivery). Their familial history was negative for congenital malformations or intellectual disability. The mother had prenatal care, registering a cleft lip and palate at 26 wk of gestation. The proband was born by cesarean section at 38 wk of gestation with a weight of 3035 g (25thpercentile), a length of 50 cm (25th-50thpercentile), an OFC of 33 cm (10thpercentile), and Apgar scores of 81and 95. She did not require neonatal management.
Personal and family history
Their familial history was negative for congenital malformations or intellectual disability.
Physical examination
Upon physical examination, her weight was 9.2 kg (< 3rdpercentile), her length was 87 cm (3rd-10thpercentile), and her OFC was 46 cm (< 3rdpercentile). She had microcephaly, upward-slanting palpebral fissures, a depressed nasal bridge, a bulbous nose, and a bilateral cleft lip and palate (Figure 1A).
Laboratory examinations
Blood, urine, and thyroid profile analyses were normal. The karyotype was 46, XX.
Imaging examinations
The abdominal ultrasound was normal. The brain magnetic resonance imaging showed cortical atrophy, pachygyria, microgyria and band heterotopia (Figure 1B).
MULTIDISCIPLINARY EXPERT CONSULTATION
The auditory study detected neurosensory hearing loss. The ophthalmic and cardiological assessments were normal.
Deoxyribonucleic acid analysis Cytoscan High Definition Array
Genomic deoxyribonucleic acid from the proband and her parents was isolated from peripheral blood samples using the Gentra Pure Gene Blood Kit and Qiagen extraction kits. The oligonucleotide-single nucleotide polymorphism (SNP) array analysis with the GeneChip Human Cytoscan high definition was carried out for the patient and her parents following the provided protocol (Affymetrix, Santa Clara, Calif., United States)and using the Affymetrix GeneChip Scanner 3000 7G. The data were analyzed using GTYPE (GeneChip Genotyping Analysis Software, version 1.0.12) to detect copy number aberrations. The resolution of this procedure was estimated at 1.15 kb with 2.67 million probes. Copy number variation (CNV) breakpoints were determined by inspecting the log2intensity ratios of SNPs within and flanking the detected regions of gain or loss.
Clinical interpretation
The interpretation of the clinical significance of all the observed CNVs was compared with the database of genomic variants (https://projects.tcag.ca/variation/), the University of California Santa Cruz genome browser (http://genome.ucsc.edu/),Ensembl Resources, Online Mendelian Inheritance of human (OMIM), ClinGen, and ClinVar. The gene content of the CNVs of interest was determined with the University of California Santa Cruz University of California Santa Cruz browser based on the reference of the human genome national center of biotechnology information build 38(hg38). For putative candidate regions containing at least one gene, the assessment included searches for similar cases in DECIPHER (https://decipher.sanger.ac.uk/)and a review in PubMed (http://www.ncbi.nlm.nih.gov/pubmed/). The CNV pathogenicity was assessed using the described guidelines[13,14]. The interpretation depended on whether a given CNV overlapped with a known genomic disorder or was present in a patient with a similar phenotype or who was reported on the database of genomic variants database.
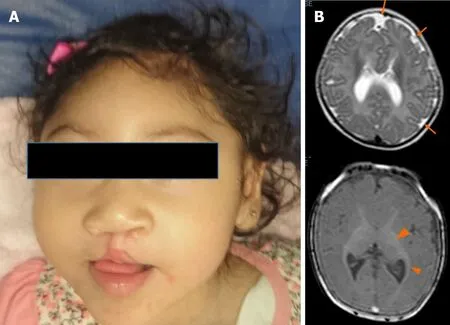
Figure 1 Patient at 1-year-old. A: Microcephaly, upward-slanting palpebral fissures, depressed nasal bridge, bulbous nose, bilateral cleft lip, and palate are showed; B: The brain magnetic resonance scan (at five months) shows cortical atrophy, simplified gyral cortical patterns (orange arrow) and band heterotopia (arrow ahead).
FINAL DIAGNOSIS
Submicroscopic 11p13 deletion involving theELP4gene with loss of heterozygosity in Xq25-26.3.
TREATMENT
There is no specific treatment for the deletion of theELP4gene, the patient was managed with rehabilitation and surgery for cleft lip and palate.
OUTCOME AND FOLLOW-UP
No improvement or progress was observed as a result of the rehabilitation.
DISCUSSION
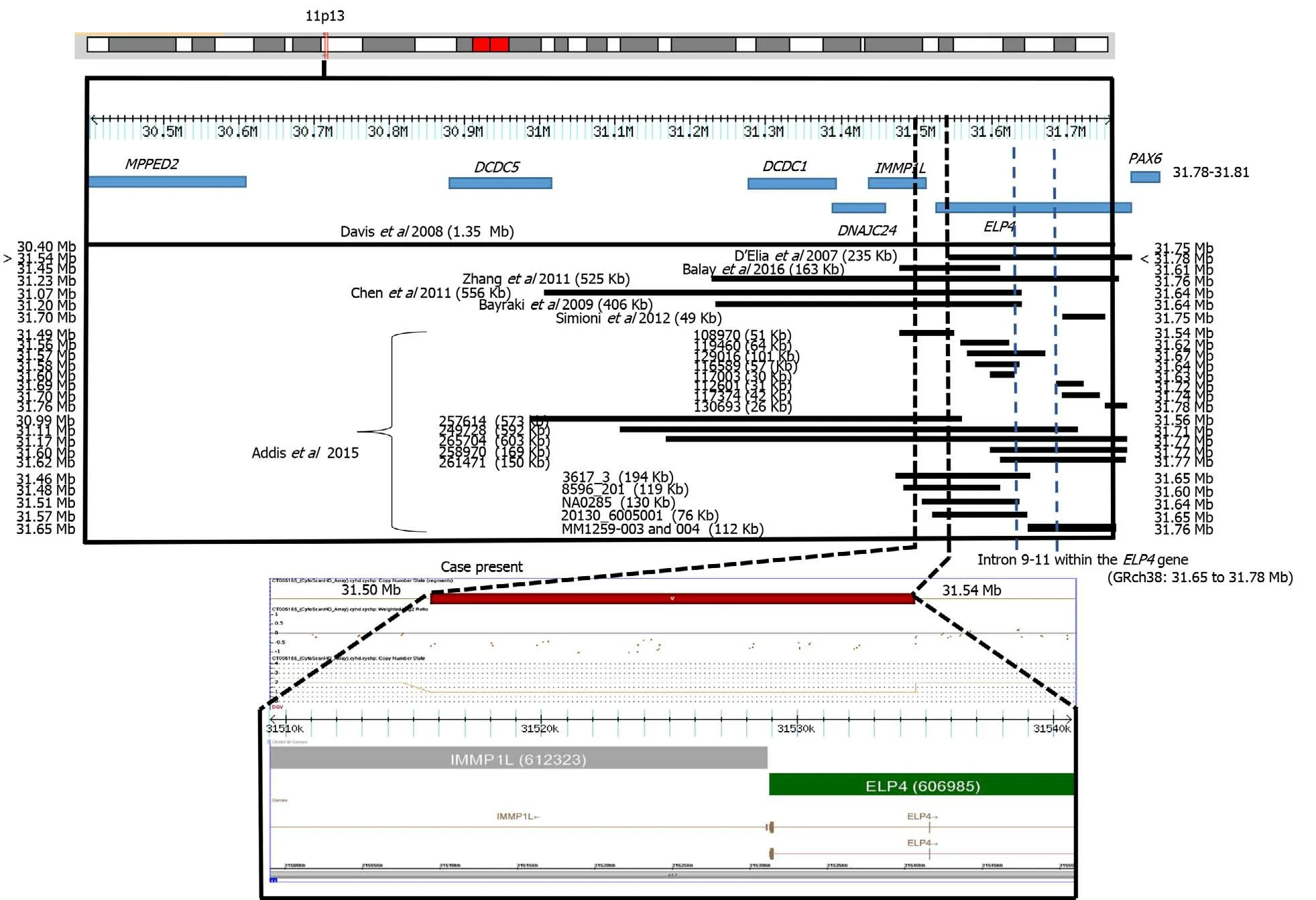
Figure 2 CytoScan high definition array and schematic representation of the result. The figure shows chromosome 11p13 and the relative positions of the MPPED2, DCDC5, DCDC1, DNAJC24, IMMP1L, ELP4, and PAX6 genes within the deleted interval. A partial molecular karyotype of the submicroscopic on chromosome 11 detected with the CytoScan high definition array is also illustrated. A single copy of the 40 Kb region was identified on log2 ratio analysis. Some affected patients with deletions in the 11p13 region are also shown.
In many patients, the clinical presentation is not fully consistent with the syndrome under consideration and laboratory confirmation rates are low. In this study, we describe the use of molecular karyotype by SNP high definition arrays to investigate the presence of pathogenic CNV in a patient with several findings consistent with a syndrome of unknown etiology. A submicroscopic deletion of 31 kb was found to be present. This CNV was located in a region with recurrent submicroscopic deletions involving theELP4gene and subjacent regions (Figure 2). TheELP4gene has been associated with intellectual disability, language development failure, autism spectrum disorder, epilepsy, and aniridia[3-9]. Our patient presented intellectual disability and language development failure but not autism spectrum disorder, epilepsy, or aniridia.These clinical data resemble those reported in a previous case withELP4gene deletion without aniridia. In this previous case, the family had a submicroscopic deletion of 163 kb in theELP4gene due to a pericentric inversion of chromosome 11p13. The patient presented intellectual disability, speech abnormalities, and autistic behaviors[10]. In another report, two families with bilateral aniridia, cataracts, and glaucoma had a deletion of 235 kb in theELP4gene[3]. Daviset al[4]also reported the case of a patient with aniridia, autism, and intellectual disability due to a 1354 kb deletion involving theELP4gene, whereas Bayrakliet al[5], studied a family with isolated aniridia that showed a deletion of 406 kb involving theELP4gene. A study in various members of a family with aniridia showed a 566 kb deletion involving theELP4gene[6], and Zhanget al[7]described a family with bilateral aniridia and congenital cataracts but without intellectual disability or other abnormalities due to a 525 kb deletion involving theELP4gene. Simioniet al[8]reported on a child with developmental delay, bilateral strabismus, aniridia, and nystagmus. The propositus showed a deletion of 49 kb in theELP4gene. Finally, Addiset al[9]made a comparison between 7235 cases and 11252 controls. The cases had language impairment, developmental delay, autism, and epilepsy. Thirteen cases presented submicroscopic deletions in 11p13. These researchers[9]reviewed DECIPHER and found 5 other cases with deletion, all overlapping with theELP4gene and suggesting a strong association between these deletions and neurodevelopmental disorders. All of these previous cases had deletions with a minimum distance of 10 kb and a maximum of 240 kb from the most proximal breakpoint to the 3′ end of thePAX6gene, with an average of 103 kb (Figure 2).
Aniridia 2 is caused by mutations affecting a long-range cis-regulatory enhancer element ofPAX6expression inside of an ultra-conserved large intronic region between exons 9 to 12 of theELP4gene, located 25 to 150 kb downstream of thePAX6gene[11].These variants inside theELP4gene do not alter its normal expression and function[12].The deletion in our patient did not include this region. Probably for this reason, the patient did not present aniridia, similar to the case of Balayet al[10]. Interestingly, the phenotypes of microcephaly, facial dysmorphia, cleft lip/ palate, neuromigration defect, and intellectual disability of our patient has been observed in the Baraitser-Winter cerebrofrontofacial syndrome 1 and 2 [OMIM 243310; BRWS1, 614583;BRWS2)][15], but theACTBandACTG1genes of Baraitser-Winter cerebrofrontofacial syndrome 1 and Baraitser-Winter cerebrofrontofacial syndrome 2 were not involved in the deletion or duplication regions or loss of heterozygosity (LOH) of our patient. In a study of 117 cases with mental delay and/or congenital malformations, 434 CNVs (195 Losses and 239 gains), including 18 pathogenic and 9 potentially pathogenic, were found to be present. Interestingly, two patients with thrombocytopenia-absent-radius syndrome were not suspected by the clinicians, possibly because of the presence of atypical features. Two patients showed a pathogenic CNV with a syndrome that was neither manifesting nor suspected, demonstrating the difficulty in making accurate clinical diagnoses in some patients with classic microdeletion and microduplication syndromes and exemplifying an unexpected discovery of non-penetrant or presymptomatic conditions. In the aforementioned study, segmental regions of loss of heterozygosity larger than 5 Mb were found in 5 patients. An analysis of microsatellite markers within the segments of LOH was carried out in some cases, confirming homozygosity of biparental origin for these regions[16].
In our patient, the SNP microarray analysis also detected a segment of LOH of 9.4 Mb, which included 39 OMIM genes, four of them have been associated with intellectual disability,OCRL, AIFM1, PHF6andHPRT1genes (HP: 0001249 in OMIM)(Figure 3). The Borjeson-Forssman-Lehman syndrome syndrome has been associated with cleft lip/palate, microcephaly, band heteropia and simplified gyral cortical patterns as the case of some female due to a de novo intragenic duplication ofPFH6[17,18], characteristics found in our patient.
Any pathogenic recessive mutation within these regions could theoretically be clinically significant, and the likelihood of this occurring would increase with the number of regions present. However, as normal genome variations, it is unlikely that most of these regions are clinically significant. A previous study with 8 families identified long segments (from 2.2% to 4.4% of the autosomal genome) and short homozygosity using short tandem repeat markers, indicating that the parents were relatively related[19]. Another study with 209 unrelated individuals from a HapMap population detected 1393 segments over 1 Mb in length (the longest length was a region of 17.9 Mb). In the aforementioned study, the subjects had an average of 6.6 homozygous segments with larger long tracts in regions of linkage disequilibrium and low recombination. This suggests that multiple ancestral megabase haplotypes persist in non-inbred human populations in broad genomic regions with below-average recombination rates[20]. Finally, an analysis of 276 neurologically normal elderly subjects among North American Caucasians using the SNP of the entire genome revealed contiguous tracts of > 5 Mb in 9.5% (26/272) of these individuals, indicating that the long homozygous segments represent segments of autozygosity due to the mating of closely related individuals, while short segments were due to linkage disequilibrium or past ancestors who were subject to distant inbreeding[21].
Microsatellite analysis in our patient’s parents could confirm the homozygosity of the biparental origin for these regions. It is important to mention that in women the X inactivation mechanism compensates for these types of molecular changes,minimizing or nullifying the phenotypic effects. To interpret LOH, it is necessary to add a catalog of new pathogenic or potentially pathogenic loci in high-quality database. This could provide the opportunity to perform genotype-phenotype correlations in a suitable number of individuals with congenital malformations and/or intellectual disability.
CONCLUSION
In conclusion, we describe a second case of deletion of theELP4gene, which confirms its association with the absence of aniridia and the presence of neurological manifestations. We also showed additional clinical data on cleft palate/Lip or facial dysmorphism and changes at the brain level, probably due to the affectation of the genes involved in the LOH region. More case reports or large studies involving patients with theELP4gene are necessary.
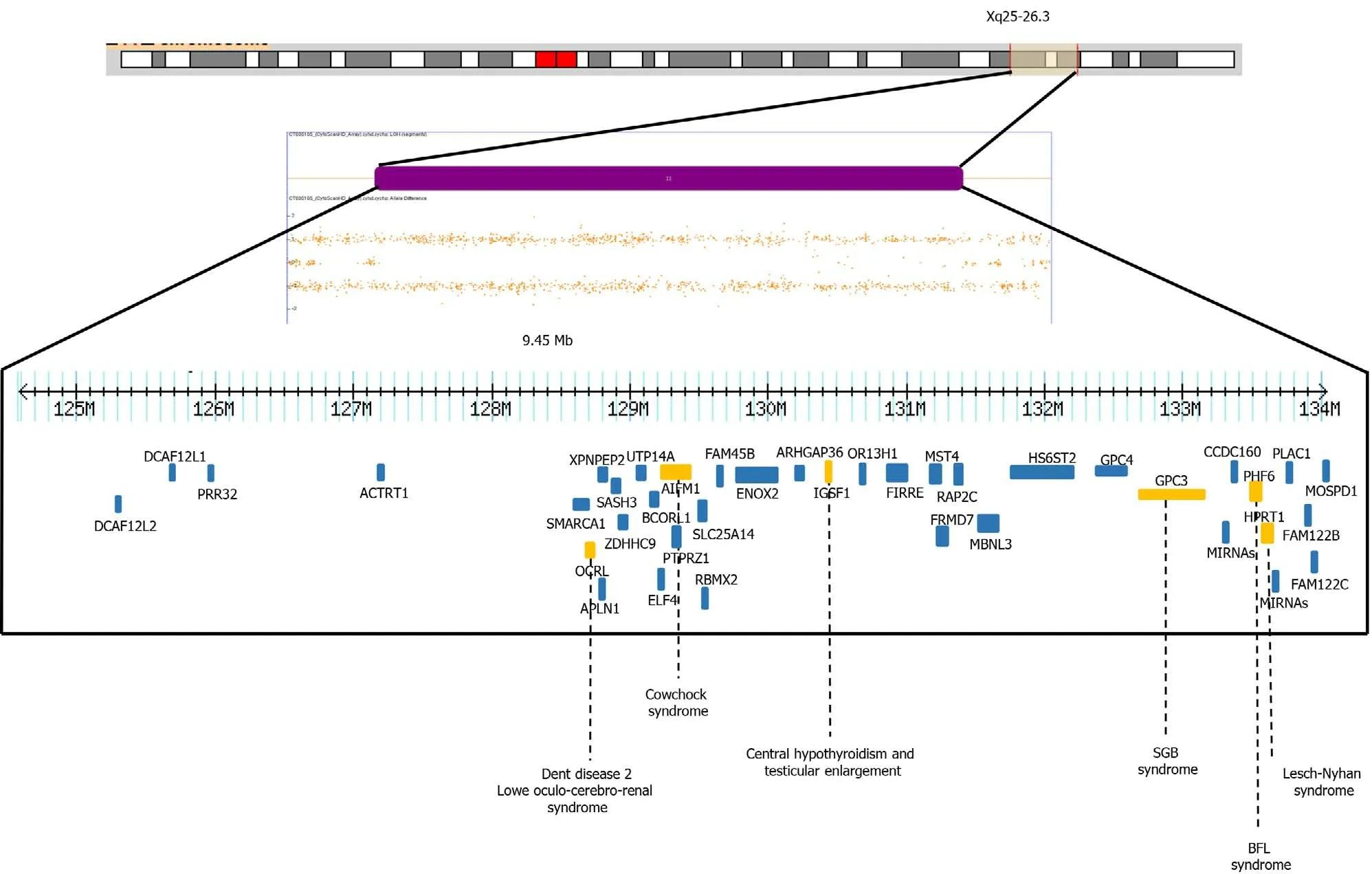
Figure 3 Depiction of allele peaks on chromosome Xq25-q26 shows homozygous (top and bottom bands) and heterozygous (middle band) allele peak bands. Note the loss of the middle band showing loss of heterozygosity of Xq25-26.3. The genes related to X-linked diseases (OCRL, AIFM1,IGSF1, GPC3, PHF6, HPRT1) (orange) such as Dent disease 2, Lowe Oculo-Cerebro-Renal syndrome, cowchock syndrome, central hypothyroidism and testicular enlargement, Simpson-Golabi-Behmel syndrome, Borjeson-Forssman-Lehman syndrome and Lesch-Nyhan are shown.
ACKNOWLEDGEMENTS
The authors thank the family for participating in this study.
杂志排行

World Journal of Clinical Cases的其它文章
- Strategies and challenges in the treatment of chronic venous leg ulcers
- Peripheral nerve tumors of the hand: Clinical features, diagnosis,and treatment
- Treatment strategies for gastric cancer during the COVID-19 pandemic
- Oncological impact of different distal ureter managements during radical nephroureterectomy for primary upper urinary tract urothelial carcinoma
- Clinical characteristics and survival of patients with normal-sized ovarian carcinoma syndrome: Retrospective analysis of a single institution 10-year experiment
- Assessment of load-sharing thoracolumbar injury: A modified scoring system