基于长短期记忆网络的抗癌肽的预测
2020-03-26孙福振李彩虹
方 春,孙福振,李彩虹,宋 莉
(山东理工大学 计算机科学与技术学院, 山东 淄博 255049)
抗癌肽(Anti-cancer peptides, ACPs)是一类小分子多肽,通常由10~50个氨基酸残基组成。它们广泛存在于昆虫、哺乳动物、植物、微生物中。例如,海鞘中的缩酚环肽、牛乳中的牛乳铁蛋白肽(LfcinB)、蚕蛹中的天蚕素B(Cecropin B)等均被发现具有抗肿瘤和抗病毒的作用[1]。抗癌肽能破坏癌细胞膜的结构,抑制癌细胞的增殖、生长,并诱导癌细胞凋亡[2]。与传统化疗药物相比,抗癌肽治疗剂具有不伤正常细胞、安全有效、成本低等优势,已经越来越受到人们的重视,成为了新药研究的热点。
后基因组时代的蛋白质序列数据爆发式地增长,迫切需要开发出自动的计算机方法,以从海量候选生物序列中快速、准确地筛选新的抗癌肽。Tyagi等[3]采用了氨基酸组成、二肽组成、氨基酸N端和C端的组成差异等信息,结合支持向量机模型进行抗癌肽的预测。Chen等[4]利用氨基酸二肽组成和伪氨基酸组分信息作为特征,结合支持向量机构建了抗癌肽的预测算法。Wei等[5]采用了氨基酸组成、二肽组成、十组氨基酸理化性质以及每种氨基酸在序列中的出现频率等信息,结合支持向量机构建了40个子模型,再以40个子模型的输出再次作为输入来搭建模型进行抗癌肽的预测。近年来,还出现了集成的机器学习方法,即整合多个或者多种机器学习算法来进行各类肽序列的预测[6-8]。这些方法的特征抽取过程繁琐,算法设计复杂,并且其性能在很大程度上依赖于特征种类数目或子模型的数量,因此,迫切需要设计出高效简单的单模型方法。
近年来,自然语言处理技术得到了突飞猛进的发展,很多关键问题的突破得益于词嵌入(word embedding)表示方法[9]。词嵌入是一种表征学习技术,可以把一个维数为所有词的数量的高维空间嵌入到一个维数低得多的连续向量空间中,每个单词被表征为实数域上的向量,并且具有相近意思的词在向量空间中的位置也靠近。生物序列本质上也可以看作是一种自然语言,每个氨基酸字母可以看作是一个单词,其性质可以看作是词意,所以,词嵌入表示方法也能被用来表示生物序列。
深度学习中的长短期记忆网络(Long Short Term Memory networks, LSTM)[10],是一种递归神经网络,近年来发展迅猛,已被证明是处理序列相关任务的利器,在一系列自然语言处理,比如,机器翻译、文档摘要、语音识别、情感分析等任务中应用广泛。本文试图将词嵌入技术与LSTM相结合来构建模型对抗癌肽进行预测,以发挥各自在序列表征和机器学习上的优势。把序列当作自然语言来处理。训练集中的序列则被视为语料库,每个氨基酸字符被当作一个单词。然后采用软件工具包将序列中的20个常见氨基酸字符映射到词嵌入空间,使每个字符对应一个多维的数值向量,继而使序列被表示成向量矩阵来作为LSTM模型的输入。最后,LSTM模型自动完成特征抽取、机器学习和预测。
1 数据与方法
1.1 数据集
本文实验所使用的数据集来自文献[5],该数据集中的样本分别收集于文献[3-4,11]。其中,正样本ACPs为均已被物理实验证实的抗癌肽,负样本数据选自于抗微生物肽中没有抗癌活性的肽。训练集ACPs500由250条抗癌肽和250条非抗癌肽构成。测试集ACPs164由82条抗癌肽和82条非抗癌肽序列构成。所有样本序列经过CD-HIT[12]过滤掉相似度高于90%的冗余序列,使训练集和测试集中的序列均不相同。数据集的详细情况见表1。
表1 数据集的统计
Tab.1 Summary of the data set

数据集序列条数正样本数负样本数训练集ACPs500500250250测试集ACPs1641648282
1.2 抗癌肽序列的长度分布
训练集中肽序列的长度分布统计如图1所示。序列长度分布在11到97个残基之间,样本的平均长度为27.12个残基。因此,在训练词嵌入向量步骤中,对序列进行整数编码时,每个序列被填充或者截断为接近平均长度的固定值30。

图1 ACPs序列的长度分布Fig.1 Length distribution of ACPs sequences
1.3 序列表征
本算法不需要人工抽取任何氨基酸理化性质,也不需要计算氨基酸组成特征等来表征肽序列,仅采用肽的字符序列作为输入,由词嵌入训练软件包首先将肽序列进行分词,即将序列切分成单个的字符;接着对序列进行固定长度的整数编码,不够设定长度的序列在末尾位置补0,超过设定长度的序列被截断,让每个字符对应到一个整数;然后通过神经网络进行词嵌入训练,将20种氨基酸字母映射到词嵌入向量空间,使得每个字符对应于一个向量表示。例如,假设整数编码的序列长度为3,而想要创建4维的词嵌入向量空间,则序列AB可转换为AB[3, 1, 0][[0.35, -0.18,-0.41,-0.21], [0.02, -0.30,-0.12, 0.40], [0.32, -0.13,-0.23, -0.15]]。以上步骤可以由Keras提供的Tokenizer API来自动完成。这样,每个序列可编码为M×N矩阵,M为设定的序列长度,N为设定的嵌入空间的向量维度。
1.4 LSTM模型
LSTM是一种特殊结构的循环神经网络,是解决长序列依赖问题的有效技术。 它由一组带有记忆功能的单元模块组成,每个单元模块由输入门、遗忘门和输出门三个门控运算来实现信息的输入、过滤和输出。这些门控运算使得LSTM能够自动提取和学习序列中对总体分类任务有用的长程相关性信息,而基于序列信息的抗癌肽的预测正好符合这类序列分类问题的特征,即序列内部隐藏着类别信息,所以,LSTM适用于肽序列的分类问题。
1.5 算法流程
本文提出的算法整体流程如图2所示。首先,由Keras提供的Tokenizer API将序列中出现的氨基酸字符通过神经网络训练后自动映射到嵌入向量空间,使每个氨基酸字符对应到一个向量表示;接着,每个肽序列可以表示成对应的数值矩阵;最后,将序列对应的数值矩阵作为LSTM模型的输入,模型自动训练、学习,并输出预测结果。这种架构将特征提取步骤和机器学习步骤交织在一起进行,可以避免将两个步骤分步执行时存在的潜在不匹配效应。
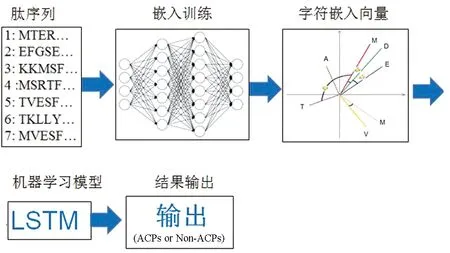
图2 算法的整体流程Fig.2 The overall flow chart of the algorithm
1.6 评价方法及评价指标
采用5交叉验证和独立测试数据集来验证本文的算法性能。 5交叉验证将训练集数据平均分成五个子集,每次采用一个子集作为验证的测试集,其余四组合并起来作为训练集。 这个过程重复进行5次,直到每个子集至少被当作一次测试集。 同时,本文也采用了独立的测试数据集来验证算法的性能。算法的评价指标包括:1)真阳性率(True Positive Rate, TPR); 2)假阳性率(False Positive Rate, FPR); 3) 正确指数;4) ROC曲线及其下的面积值AUC。各指标计算公式如下:
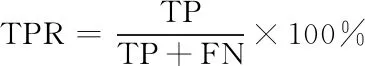
(1)
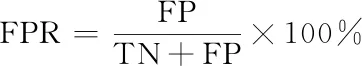
(2)
正确指数=(TPR+1-FPR)/2
(3)
式中:TP指被预测为正的正样本数;FP指被预测为正的负样本数;TN指被预测为负的负样本数;FN指被预测为负的正样本数。
2 实验与结果分析
2.1 模型参数优化
因为预测抗癌肽实质是二分类问题,即判别输入的肽序列是否为抗癌肽,所以,在编译模型时,损失函数设置为适合二分类的 binary_crossentropy,优化函数则为rmsprop,激活函数为sigmoid。
为了得到合适的词嵌入向量空间的维度,本文分析了不同维度设置下模型的性能。设定LSTM隐藏层存储单元的个数为100,词嵌入向量空间的维度分别为10、20、30、40、50、60、70、80和90,在训练集上采用5交叉验证时模型所对应的ROC曲线如图3所示。当嵌入维度在80时,取得的ROC曲线下面积值最大(灰色线所示),即预测性能最好,所以,本文最终设定词嵌入空间的向量维度为80。

图3 不同词嵌入维度对应的ROC曲线Fig.3 The ROC plots of the predictor with different number of embedding dimensions
接着为了得到合适的LSTM隐藏层存储单元的个数,本文分别设置存储单元数为20、40、60、80、100、120、140、160和180,模型在训练集上进行5交叉验证时,所对应的ROC曲线如图4所示。 当存储单元数为140时,模型性能最好(黄色线所示)。因此,本文设定LSTM隐藏层存储单元的个数为140。由于抗癌肽样本的平均序列长度仅为27个残基,在进行词嵌入训练时,序列被填充或截断到固定长度30,因为长度比较短,所以本文只使用了单层LSTM网络。最后,得到的预测模型中各层的输入和输出参数情况如图5所示。其中,(None,30)和(None,30,80)分别表示对应层输入是30维,输出是一个30×80维的矩阵,None表示此处不包括处理数据的批量维度。

图4 不同LSTM存储单元对应的ROC曲线Fig.4 The ROC plots of the predictor with different number of memory units

图5 预测模型中每层的参数情况Fig.5 Parameters of each layer in the predictor
2.2 训练集和测试集上的准确率和损失函数分布
设置训练参数epoch为200次时,模型在训练集和独立测试集上的准确率和损失函数的曲线如图6所示。从图6可以看出,整个训练过程中的准确率和损失函数在训练集和测试集上的曲线相互靠近,说明训练过程中没有发生过拟合现象,即模型具有较好的泛化能力。当该模型在独立测试集ACPs164上进行验证时,所对应的ROC曲线如图7所示,该方法的 TPR值达到0.756, AUC值达到0.895。
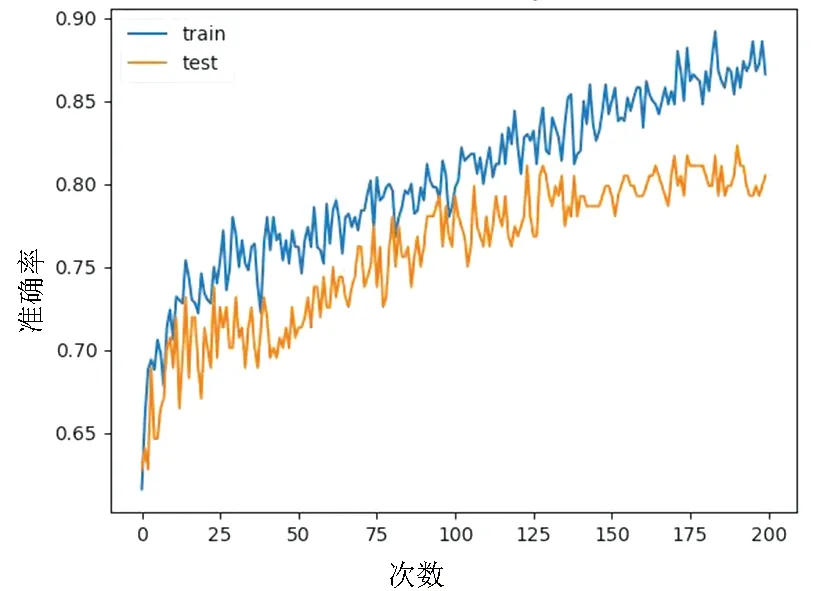
(a)准确率

(b)损失函数图6 模型准确率曲线和损失函数曲线Fig.6 The accuracy plot and loss function plot of the model

图7 模型在独立测试集上的ROC曲线Fig.7 ROC plot of proposed method tested on the test dataset
值得注意的是,本文方法的性能虽然略逊于ACPred-FL[6]预测方法的性能,但此方法是基于单模型的方法,整个预测过程仅使用了一个机器学习模型,没有采用任何其他分类器的结果再次作为输入。而ACPred-FL[6]是基于组合的机器学习方法,它包括了两层的支持向量机,其第二层组合了第一层中40个支持向量机子模型的结果,因此,其性能很大程度上依赖于子模型的数量,而一般组合的子模型越多,性能会越好,但计算成本也随之急剧增加。
2.3 不同的固定序列长度设定对预测结果的影响
本文也分析了对序列进行固定编码长度设置时,不同的设定长度对最终预测性能的影响。当设定的固定长度分别为10、20、30、40和50时,训练后的模型在独立测试集上的ROC曲线如图8所示,其中固定长度为30个残基时的模型性能最佳。其可能的原因是,抗癌肽通常由10~50个氨基酸残基组成,且平均长度接近于30,所以,当填充长度设定在平均长度左右时,需要填充或者截断的序列数量最少,这样可以最大程度上保留数据集中肽序列的原始特征信息。

图8 不同序列填充长度ROC曲线Fig.8 ROC plots with different sequence padded length for the integer encoding
2.4 与其他两种算法的比较
目前有很多研究表明,将深度学习中热门的两大模型——卷积神经网络(Convolutional Neural Network, CNN)和LSTM联合在一起使用可以取得更好的效果。本文也尝试将基于CNN模型和基于CNN-LSTM联合模型的方法用于抗癌肽的预测,并将这两种方法的结果与本文的进行了比较。此处,CNN为单层的一维卷积神经网络,卷积层包含32个大小为3的卷积核,激活函数采用relu,池化层核大小为3,丢失率为0.5。CNN-LSTM的联合模型中的参数与单独的CNN模型和LSTM模型中对应位置的参数均相同。三种方法在独立测试集上的ROC曲线如图9所示,各项指标见表2。图9和表2显示,本文基于LSTM的方法性能最好,正确指数达到0.829,AUC值达到0.895;基于CNN的方法性能次之;而基于CNN-LSTM联合模型的方法性能最差。结果表明在本文加入CNN 模块并不能带来预测性能的改进。其可能的原因是,抗癌肽序列的平均长度比较短, 使用LSTM足以捕捉到序列中对整体分类任务有用的关键的信息,而加入CNN模块后反而加重了噪音特征的混入。
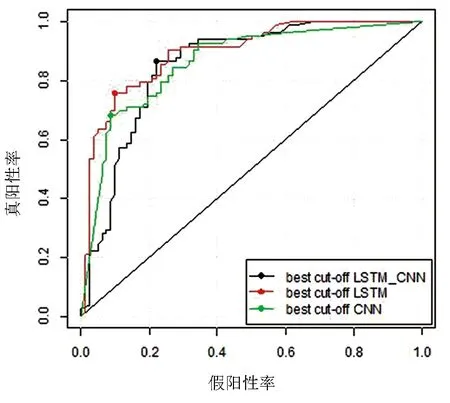
图9 三种方法在独立测试集上的ROC曲线Fig.9 ROC plots of three different architecture-based methods applied on the test dataset
表2 三种方法在独立测试集上的性能比较
Tab.2 Performance comparison of three different methods on test sets

方法正确指数真阳性率假阳性率AUC基于CNN的方法0.7990.6830.0850.862基于CNN-LSTM的方法0.8230.8660.2200.854基于LSTM的方法0.8290.7560.0980.895
3 结论
本文提出了一种基于词嵌入的序列表示方法与LSTM模型相结合的算法,进行抗癌肽的预测。该算法具有如下特点:
1) 实现了较好的预测性能,在测试集上进行验证时,该方法正确指数达到0.829,AUC值达到0.895。
2) 本算法是基于单一模型的算法,不需要任何人工特征提取和特征重建步骤,也不用组合其他分类器的结果,从而简化了算法设计。
3)在LSTM机器学习模型中增加CNN模块或者使用CNN模型代替LSTM模型来进行预测,均不能带来算法性能的改进。
