不同环境中纤维素降解菌群多样性差异分析
2020-03-25李宪臻
杨 明,袁 悦,李宪臻,杨 帆
(大连工业大学 生物工程学院,辽宁 大连 116034)
【研究意义】纤维素是地球上最丰富的有机聚合物,平均占植物生物量的30%~50%[1-2]。研究纤维素降解相关菌群对于解决农业废弃物的回收利用、可再生能源的开发等环境和能源问题有着重要意义[3-4]。纤维素是由D-葡萄糖以β-1,4糖苷键连接构成,由于聚合度高加之结晶度高的特性,导致其难以降解,极大地限制了纤维素在上述相关领域的应用[5]。纤维素的生物降解需要多种纤维素酶协同完成,包括内切葡聚糖酶,外切葡聚糖酶,纤维二糖水解酶和β-葡糖苷酶等[6-7]。纤维素酶是由纤维素降解菌合成分泌,纤维素降解菌广泛存在于自然界中,多分布在纤维素聚集和降解的环境[8]。这些环境中多以微生物复合菌系协同作用的方式完成纤维素的降解,因此研究环境细菌多样性对于阐明纤维素生物降解机理显得尤为重要。【前人研究进展】瘤胃、堆肥和森林土壤等是研究的较多的纤维素降解环境[9],其中含有大量能够降解纤维素的微生物。目前已有很多关于纤维素酶产生菌的研究报道,其中大多数中筛选自动物瘤胃、堆肥、森林土壤等环境[10-11]。然而,基于纯培养的传统分析方法存在很大的局限性,自然界中超过99%的微生物都是不可培养的[12]。随着高通量测序技术的出现和发展,16S rDNA基因扩增子高通量测序分析技术可直接针对细菌群落进行分析,基于16S rDNA的高变区序列信息对微生物群落中的细菌进行物种种类和丰度的鉴定[13],能够更全面地分析微生物多样性。Li等[14]应用16S rDNA基因测序方法对牛瘤胃菌群进行了分析,获得了以45个核心微生物属为主的共107个细菌属,展现出丰富的菌群多样性。Ren等[15]对牛粪堆肥中的微生物群落进行了16S r DNA基因高通量测序分析,发现其优势菌门包括拟杆菌门、变形菌门、厚壁菌门和放线菌门,并且发酵的不同阶段具有不同的细菌。王志方等[16]应用16S rDNA高通量测序技术对棉秸秆自然腐解过程中不同时间点的样品进行分析,发现在第7天时细菌群落多样性在属水平上与腐解起始阶段差异显著(P<0.05),随着腐解时间延长,菌落多样性趋于一致;在整个腐熟过程中始终存在5个优势属,功能预测为降解纤维素、木质素、果胶类物质以及提供氮素营养。【本研究切入点】近年来,测序技术以超摩尔定律的速度在不断发展,测序成本越来越低[17],16S rDNA基因测序数据也在不断加速积累,且海量的数据被公开于NCBI-SRA数据库,这其中也包括瘤胃、堆肥、土壤等不同自然环境下纤维素降解菌群数据。此外,尽管16S rDNA基因测序技术在纤维素降解菌群的结构和多样性研究方面取得了进展,但这些研究多数仅仅关注的是来自某种特定环境的纤维素降解菌群,却很少有研究对不同环境下的纤维素降解菌群进行比较分析。【拟解决的关键问题】本研究选取了来自NCBI-SRA数据库中的牛瘤胃、牛粪堆肥、森林土壤3种代表性的纤维素降解菌群和作为对照的家犬肠道微生物群落共102个的16S rDNA样本的高通量测序数据,对这些数据进行了细菌群落多样性的共性和差异分析,以评估不同来源的纤维素降解菌群在各个分类级别上的共性和差异特征,鉴定各类菌群的生物标志物,对功能主导性菌群进行挖掘,为进一步探索纤维素降解菌群的功能、研究纤维素降解生物学机制以及开发有利用价值的微生物提供参考。
1 材料与方法
1.1 试验材料
1.1.1 数据采集 16S rDNA Illumina Miseq高通量测序数据搜索自NCBI(National Center of Biotechnology Information)的SRA(Sequence Read Archive)数据库中收录的牛瘤胃BR(SRA ID:SRP156364)、牛粪堆肥CDC(SRA ID:SRP074428)、森林土壤FS(SRA ID:SRP125392)和家犬粪便DF(SRA ID:SRP092477)4个项目的样本数据。
1.1.2 数据筛选标准 将搜索到的数据进行筛选,保留16S rDNA数据筛选均需符合下列生物学标准和相应的技术标准。生物学标准:(1)牛瘤胃菌群来自爱尔兰米斯郡欧洲农业食品有限公司的健康牛瘤胃;(2)牛粪堆肥菌群来自加拿大农业与食品学会的牛粪好氧堆肥;(3)森林土壤菌群FS采集自中国辽宁省抚顺市林场的针叶林表层土(深度不超过10 cm)细菌;(4)家犬粪便DF微生物样本为未作处理的健康状态下家犬的新鲜粪便样本,来自密苏里大学(美国密苏里州哥伦比亚)。技术标准:(1)16S rDNA基因扩增序列为V4区;(2)测序平台为Illumina Miseq测序平台;(3)测序数据量不小于30 Mbp。
1.2 实验方法
应用SRA Toolkit软件中的fastq-dump命令将sra格式的文件转换为FastQ格式[18]。FastQ格式的双末端reads经过滤获得高质量reads[19]。应用FLASH软件[20]将双末端reads拼接成Tags,并去除没有overlap关系的reads,低质量的Tags(双末端reads最小重叠长度为15 bp,其重叠区域错配率>10%)以及携带的引物序列的Tags,从而得到高质量Tags。应用SRA Toolkit软件中的fastq-dump命令下载SRA格式的文件并转换为FastQ格式。应用USEARCH GLOBAL方法,将Tags重新比对回OTU序列得到每个样品在每个OTU的丰度统计表[24]。应用RDP classifier(v2.2)[25]将OTU与Greengenes数据库[26]比对进行物种注释(如果Tag长度大于等于250 bp,置信度阈值设置为0.8,否则设置为0.5),去除没有注释结果和非细菌物种的OTU,并根据注释结果计算每个样本组在各分类水平(门、纲、目、科和属)的群落组成结构信息。
1.3 数据统计分析
Alpha多样性反映了单个样本内物种的多样性。mothur软件计算Alpha多样性的观察指数(observed species index)、赵氏指数(chao1 index)、艾斯指数(abundance-based coverage estimator index,ACE index)、香农-维纳指数(shannon-wiener index)和辛普森指数(simpson index)。观察指数反映的是已经检测到的物种数量;赵氏指数和艾斯指数则是基于物种和丰度值对真实物种数的推测;香农-维纳指数和辛普森指数则反映群落的多样性,是物种数量和丰度分布均一性的综合反映。Beta多样性分析用于评估样品在物种复杂性上的差异距离。通过QIIME(v1.80)[27]计算加权和非加权Unifrac距离[28],主坐标分析(PCoA)使用的是R软件(v3.0.3)的WGCNA,stats和ggplot2软件包,即将加权或非加权UniFrac距离矩阵转化为一组新的正交轴,其中最大变异的因子作为第一主坐标,第二大变异因子作为第二主坐标表示。R软件的VennDiagram包用于绘制Venn图,ggplot2包用于绘制箱线图和柱状图。Metastats软件(http://metastats.cbcb.umd.edu)用于组间显著性差异分析。统计假设检验均以α=0.05为标准界定是否具有统计学意义。LEfSe(v1.0)[29]用于发现各组中物种生物标志物(Biomarker),其LDA分数的阈值设为4。
2 结果与分析
2.1 不同纤维素降解环境菌群多样性分析与比较
牛瘤胃BR、牛粪堆肥CDC、森林土壤FS和家犬粪便DF 4个组共102个样本的16S rDNA基因扩增子测序数据经过前处理,共获得8 122 885条高质量16S rDNA标签序列,平均每个样本的数量约为79 636,在97%的相似水平上进行操作分类单元(OTU)序列聚类,共获得80 386个OTU类群。观察物种指数(observed species index)结果显示物种平均丰富度由高到低为牛粪堆肥、家犬粪便、牛瘤胃,森林土壤仅显著低于牛粪堆肥,但与其他两组无显著差异。赵氏指数(chao1 index)和艾斯指数(ACE index)结果均显示平均物种丰富度最高为牛粪堆肥,其次为家犬粪便和牛瘤胃,而森林土壤与其他环境未表现出显著差异。香农-维纳指数(shannon-wiener index)和辛普森指数(simpson index)均可以用来估算细菌多样性。香农-维纳指数值越大,细菌多样性越高;辛普森指数介于0和1之间,与香农-维纳指数相反,数值越低细菌的多样性越高。因此,这两个细菌多样性指数均显示牛粪堆肥菌群多样性显著高于其他环境,且牛瘤胃的香农-维纳指数显著高于家犬粪便。

本研究对4个群落的Unifrac Beta多样性进行分析并用主坐标分析(PCoA)的可视化方式进行展示,以评估各样本之间的相似度和差异。如图2所示,非加权Unifrac PCoA的第一主坐标贡献率达19.6%,第二主坐标贡献率达15.2%,共代表总变量的34.8%;加权Unifrac PCoA的第一主坐标贡献率达31.2%,第二主坐标贡献率达16.3%,共代表总变量的47.5%。基于OTU丰度数据可以把同一个样本组的菌群样本聚集在一定区域内,而不同样本组间获得了很好的分离。
2.2 不同纤维素降解环境菌群的组成差异
所有样本组中总共检测出55个门,其中Proteobacteria、Firmicutes、Bacteroidetes和Actinobacteria是4个最优势门,他们均在牛瘤胃和牛粪堆肥中均占有很大比例(图3a)。作为对照的家犬粪便菌群以Bacteroidetes、Firmicutes、梭杆菌门(Fusobacteria)和Proteobacteria为最优势门,Fusobacteria是其特有门,另外存在极少量的Actinobacteria。牛粪堆肥中Bacteroidetes相对丰度显著低于其他菌群(图3d),而Actinobacteria的相对丰度显著高于其他菌群(图3e)。与牛瘤胃和牛粪堆肥不同,森林土壤环境中的Proteobacteria显著高于其他纤维素降解环境(图3b),仅存在极低丰度的Firmicutes(图3c)。
属水平上所有样本中总共检测到1 936个属,选取相对丰度最高的前17个属(不包括Unclassified、Others和Uncultured)进行了比较(图4a)。家犬粪便中的优势属包括梭杆菌属(Fusobacterium)、拟杆菌属(Bacteroides)、粪杆菌属(Faecalibacterium)、Prevotella-9和乳酸菌属(Lactobacillus),除了Lactobacillus外其余均为家犬粪便中的特有菌属。牛瘤胃中相对丰度较高的属有琥珀酸弧菌UCG-001属(SuccinivibrionaceaeUCG-001)、Prevotella-7、甲烷短杆菌属(Methanobrevibacter)、Succiniclasticum、Prevotella-1和少量的Lactobacillus,其中SuccinivibrionaceaeUCG-001和Succiniclasticum仅在牛瘤胃中被观察到,而Prevotella在动物消化道来源的环境中均有发现,不同的是Prevotella-7和Prevotella-1存在于牛瘤胃中,而Prevotella-9存在于家犬粪便中。Methanobrevibacter在牛瘤胃和牛粪堆肥中均被检测到,牛粪堆肥中的相对丰度较低。牛粪堆肥中的观察到的特有菌属包括Halocella、Limnochordaceae-ge和氢孢菌属(Hydrogenispora)。另外,Pseudomonas仅在牛粪堆肥和森林土壤中被检测到,且在前者中相对丰度极显著低于后者(P<0.01)。此外,黄杆菌属(Flavobacterium)、NS3a海洋菌属(NS3amarine group)和假噬纤维菌属(Pseudarcicella)属为森林土壤中所特有。
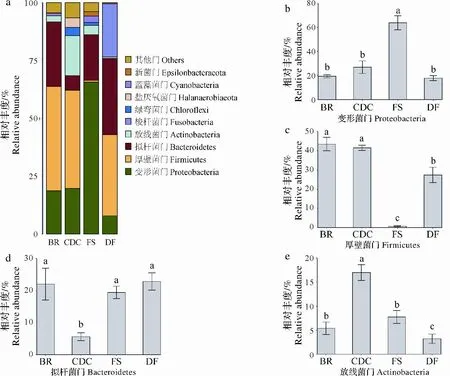
图3 不同环境菌群门水平相对丰度分布和优势菌门差异比较Fig.3 Relative abundance distribution at phylum level and the difference of relative abundance of dominant phylum in different environments
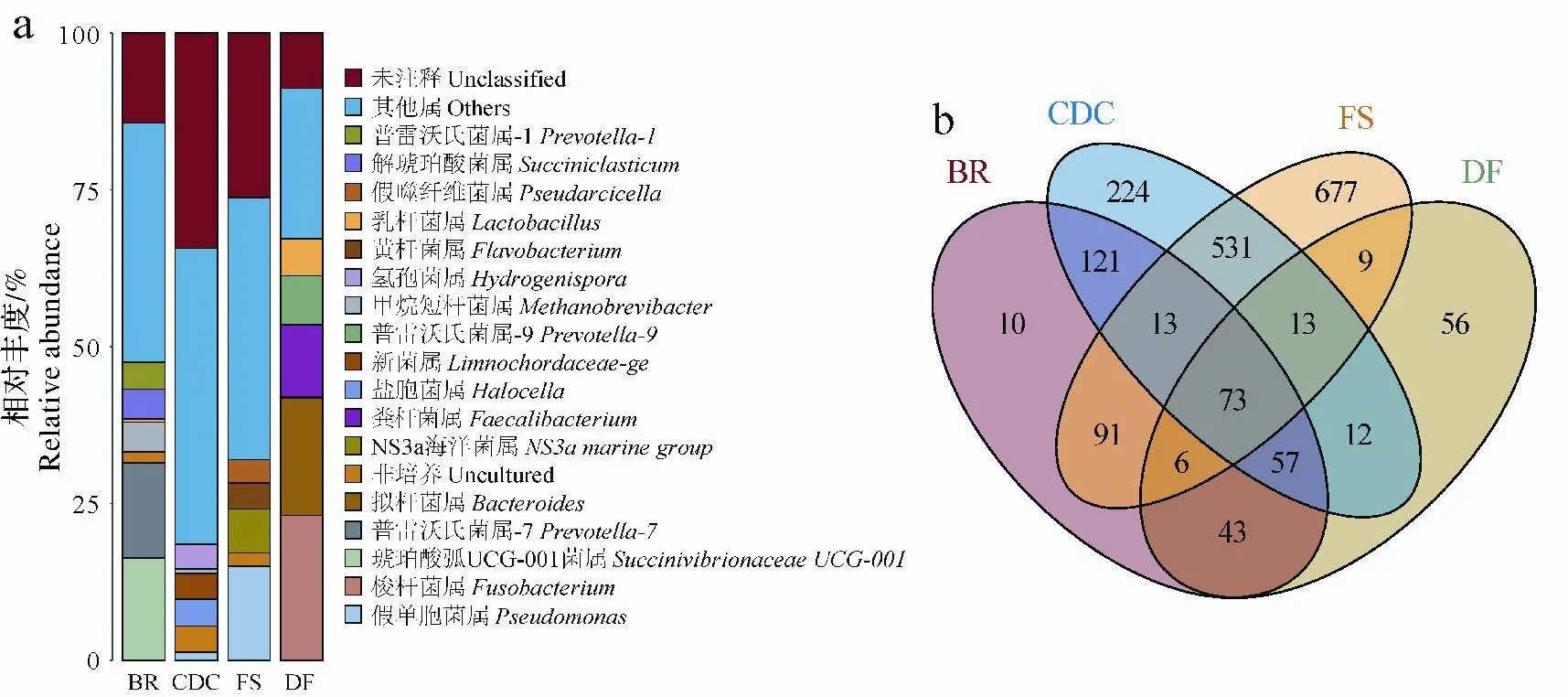
图4 不同环境菌群下属水平优势菌群相对丰度分布和Venn图Fig.4 Relative abundance distribution and Venn diagram at genus level in different environments
韦恩图分析(图4b)表明仅有73(4.44%)个属是4类菌群共有的,86个属是3类纤维素降解菌群共有的。而森林土壤和牛粪堆肥中的特有属较多,分别为677和224个;牛瘤胃和家犬粪便中的特有的属分别仅观察到10和56个。属水平统计的物种丰富度从高到低表现为森林土壤、牛粪堆肥、牛瘤胃、家犬粪便,分别检测到1 413、1 044、414和269个属。

图5 不同环境菌群的LEfSe分析Fig.5 LEfSe analysis of bacterial communities from different environments
2.3 不同纤维素降解环境菌群生物标志物分析
为了更进一步考察不同环境中纤维素降解菌的差异寻找不同环境的关键菌群,本研究对物种注释后获得的OTUs丰度谱进行了线性判别分析效应值(LEfSe)分析(图5)。LEfSe为最近出现的一种基于线性判别分析(linear discriminant analysis,LDA)效应量(effect size)的分析方法,其本质是将非参数统计检验(Kruskal-Wallis多组检验与Wilcoxon秩和检验)与线性判别分析相结合,从而筛选关键的生物标志物(比如关键功能类群/物种)[30]。结果显示,牛瘤胃、牛粪堆肥、森林土壤及家犬粪便组间具有显著差异的不同级别分类单元重要的生物标志物数量分别为12、5、9和13。由于不同分类级别的生物标志物之间存在从属关系,表1对LEfSe分析结果进行了整理,并对各个生物标志物的相对丰度进行标识,以直观展示不同环境中重要生物标志性物种及其相对丰度。生物标志物将以门水平和最低级别分类水平加以描述。对照组家犬粪便的生物标志物包括Bacteroidetes的Bacteroides、Fusobacteria的Fusobacterium以及来自Firmicutes的Faecalibacterium。与对照组同为动物消化系统直接来源菌群,牛瘤胃组中的生物标志物包括广古菌门(Euryarchaeota)、来自Proteobacteria的SuccinivibrionaceaeUCG-001、来自Bacteroidetes的Prevotella-7、来自Firmicutes的Selenomonadales和Lachnospiraceae;作为动物消化系统和体外环境来源的混合菌群,牛粪堆肥组的生物标志物为来自Actinobacteria门Actinobacteria纲和来自Firmicutes的Bacillales;作为体外环境来源菌群森林土壤组生物标志物以Proteobacteria为主,包括Pseudomonas、Alphaproteobacteria、伯克氏菌科(Burkholderiaceae)以及Bacteroidetes门的Flavobacteriaceae。

表1 不同环境菌群的生物标志物及其相对丰度*Tab.1 Relative abundance of biomarkers from different environments
3 讨论
随着二代测序技术成本逐渐降低和技术的不断更新,16S rDNA扩增子测序技术被广泛用于纤维素降解菌群多样性的研究中,加强了人们对纤维素降解菌群结构和功能的认知。然而大多数研究者仅关注于单一环境类型中的纤维素降解菌群,却鲜有对不同环境下的纤维素降解菌群进行分析。本研究以家犬粪便菌群16S rDNA序列数据为对照,选择来自牛瘤胃、牛粪堆肥、森林土壤的3类菌群数据作为不同环境条件下的纤维素降解菌群,对其菌群结构多样性进行对比分析,旨在阐明菌群的结构和功能,为进一步挖掘有潜在应用价值的纤维素降解菌以及解析纤维素降解功能奠定理论基础。
菌群多样性分析发现,牛粪堆肥菌群在物种种类和物种丰度的均一性上均高于其他组别,牛粪堆肥菌群是来自于厌氧牛体内环境和体外环境的混合菌群,这可能是造成上述现象的一个原因。森林土壤组内样本的alpha多样性指数之间差异较大,可能与其复杂多变的环境和采样的时间地点有关,菌群多样性高可能更有利于应对复杂多变环境,比如对温差、湿度等的适应性。此外,物种丰度分析显示森林土壤中属的数量高于牛粪堆肥,可能是由森林土壤组内样本差异大造成。PCoA分析结果充分说明了样本分组的可靠性,同时表明来源于不同环境的菌群之间的群落多样性存在较大差异。
本研究检测到Proteobacteria、Firmicutes、Bacteroidetes和Actinobacteria 4个来自纤维素降解菌群的优势门,这与先前的许多报道一致[31-34]。Firmicutes存在大量纤维素降解菌[35],可能是牛瘤胃和牛粪堆肥菌群中降解纤维素的主要门。Bacteroidetes对多糖等碳水化合物具有较好的降解能力[36],在4个菌群中均大量存在,是家犬肠道中的生物标志物。有研究表明,Actinobacteria是降解木质素与纤维素的主要功能菌门[2,37]。在土壤有机层中丰度最高的门为Proteobacteria和Bacteroidetes,而Firmicutes则更多的存在于矿物土壤中[38],这与本研究结果一致,说明Proteobacteria是森林土壤中纤维素降解重要门。此外,Euryarchaeota常被发现于牛瘤胃中,然而很少有证据证明其纤维素降解能力,尽管Sorokin等[39]首次盐古菌(Haloarchaea)具备降解几丁质和纤维素的能力,然而本研究中并未检出盐古菌。
LEfSe分析可帮助研究者发现生物标志物,但也存在灵敏度不足的情况导致一些特异性的功能性细菌漏检,因此对LEfSe分析结果和环境特异性属进行整合可作为一种优化的方法鉴定环境特异性纤维素降解菌。家犬粪便以Fusobacterium、Bacteroides和Faecalibacterium为生物标志物,其特有优势菌属为Prevotella-9和Lactobacillus,均为杂食性哺乳动物肠道常见菌株[40]。牛瘤胃的生物标志物包括Lachnospiraceae、Euryarchaeota、SuccinivibrionaceaeUCG-001、Prevotella-7和Selenomonadales,除此之外其特有优势菌属还包括Prevotella-1,Succiniclasticum和Methanobrevibacter。Clostridiales是瘤胃中纤维小体的主要生产者[41]。Lachnospiraceae属于Clostridiales,是牛瘤胃中的优势菌,存在能够降解纤维素的细菌,如丁酸弧菌属(Butyrivibrio)和互养球菌属(Syntrophococcus)等[42]。另外,Seshadri等[43]在410个瘤胃细菌和古细菌基因组中发现大量参与形成纤维素体的Clostridiales细菌。SuccinivibrionaceaeUCG-001是Aeromonadales中的最优势属,Succinivibrionaceae可发酵淀粉等碳水化合物产生琥珀酸和醋酸盐,有可能参与纤维素代谢中间产物的降解,但尚未发现文献报道该科可降解纤维素。Prevotella是纤维素降解过程中的优势属[44-46],Prevotella的细菌(如P.ruminicola等)可直接生产纤维素酶,厌氧环境中可与其他纤维素降解菌协同降解纤维素[35,47]。Selenomonadales来自厌氧菌纲(Negativicutes)最优势的属为月形单胞菌属(Selenomonas),以降解淀粉为主[48],但某些菌株(如S.ruminantium)具备纤维素降解功能[49]。Succiniclastium是典型的纤维降解菌,能发酵降解纤维素或纤维二糖产生琥珀酸、乙酸和二氧化碳等[50]。Methanobrevibacter是Euryarchaeota门的最优势属,是一类产甲烷菌[51]。牛粪堆肥中的生物标志物为Actinobacteria和Bacillales。Bacillales中存在较多纤维素降解菌,比如常见的来源于堆肥或土壤纤维素降解菌Bacillus等[52]。Actinobacteria(纲)有着基因组GC含量高以及细胞壁厚的特点,能适应高温、强碱等极端环境,其中很多细菌具有纤维素降解能力,如Actinanyces cellulose、诺卡氏菌属(Nocardia)和链霉菌属(Streptomyces)等[53-55],因此Actinobacteria可能是牛粪堆肥中重要的纤维素降解菌。另外,牛粪堆肥中特有优势菌属包括Halocella、Limnochordaceae-ge和Hydrogenispora,目前已经发现的Halocella包括H.cellulosilytica和H.sp.SP3-1,可在高盐环境下生存,均具备降解纤维素和纤维二糖的能力[56-57]。Hydrogenispora来自梭菌纲(Clostridia),与Clostridiumsp.6-31较为近缘,能够发酵降解多种碳水化合物[58],但尚无充分证据支持其纤维素降解能力。有关Limnochordaceae-ge属的相关报道并不多,有待于进一步研究。森林土壤组的生物标志物包括Alphaproteobacteria、Pseudomonas、Flavobacteriaceae以及Burkholderiaceae。Pseudomonas属于严格好氧型纤维素降解菌,可产生纤维素酶[59-60],在牛粪堆肥中的丰度低于森林土壤,可能是森林土壤和牛粪堆肥的重要的纤维素降解菌。Flavobacteriaceae的优势属为NS3amarine group和Flavobacterium,仅发现于森林土壤。Flavobacterium能够降解复杂有机物质如纤维素、半纤维素、几丁质等有机聚合物[61-63]。NS3amarine group常常在海洋藻类中与Flavobacterium一起被发现[64],推测其可能具备纤维素降解功能。Alphaproteobacteria在森林土壤中被大量发现,最优势属包括Brevundimonas、生丝单胞菌属(Hyphomonas)和紫杆菌属(Porphyrobacter)等,物种种类较为丰富,但各属的相对丰度较低,但其中也存在纤维素降解菌,比如Rhodobacterales中Brevundimonas和Hyphomonas均具备降解纤维素能力,其中Brevundimonas可在低温环境下产生纤维素酶[65-66]。森林土壤样本中的Burkholderiaceae的诸多细菌具备降解纤维素的能力如伯克氏菌属(Burkholderia)[67]。Pseudarcicella属是2012年Kämpfer等[68]在医用水蛭皮肤上发现的一个新属,被归属于噬纤维菌科(Cytophagaceae),其中绝大多数物种均有纤维素降解功能[69],故推测其具备纤维素降解功能。
4 结论
本研究对4组来自于不同环境的共102个环境微生物群落样本数据进行了分析。Alpha多样性显示,牛粪堆肥中物种种类和丰度的alpha多样性指数显著高于其他两种纤维素降解菌群。基于非加权和加权Unifrac距离的PCoA分析说明不同环境中的纤维素降解菌群的微生物多样性差异较大。Proteobacteria、Firmicutes、Bacteroidetes和Actinobacteria是本研究在各类环境菌群间检测到的最具优势的4个门,不同环境间在门水平就存在着显著差异,表现出了特异性。各类环境菌群间在属水平上差异极大,绝大多数属表现出有和无的定性差异,进一步说明不同样本的纤维素降解菌群多样性的不同。LEfSe分析鉴定到了牛瘤胃、牛粪堆肥、森林土壤中的生物标志物分别有5、2和4个。最后,LEfSe结果、属水平相对丰度信息以及先前的文献报道的综合分析和讨论,鉴定到牛瘤胃、牛粪堆肥、森林土壤中同时具备环境特异性和纤维素降解功能的细菌分类单元分别有4,3和5个。本研究从宏观层面即来自不同环境的纤维素降解菌群的多样性和差异进行了分析,为进一步阐明天然细菌群落的纤维素降解的协同作用机理奠定了基础,同时为构建具备相应环境适应性的纤维素降解菌群以及筛选在生产生活中有应用价值的菌株提供了理论指导。
