气相色谱-质谱法测定纺织品中喹啉与异喹啉
2020-03-19高永刚牛增元叶曦雯张艳艳连素梅
高永刚,牛增元*,罗 忻,叶曦雯,张艳艳,连素梅
(1.青岛海关技术中心,山东 青岛 266003;2.青岛市产品质量监督检验研究院,山东 青岛 266022;3.石家庄海关技术中心,河北 石家庄 050000)
喹啉又名苯并吡啶,系含氮(杂)双环芳烃,是一种重要的有机合成原料[1],其异构体为异喹啉。喹啉、异喹啉是一类重要的生物碱,具有生理活性,被广泛用于医药、农药、精细化工等领域。在纺织及印染行业,喹啉、异喹啉衍生物主要用于合成C.I.酸性黄3、C.I.直接黄22、C.I.溶剂黄33和Palanil黄3G等黄色染料。此外,喹啉经硝化、还原得到的氨基喹啉主要用于纺织品染色助剂。喹啉为危险化学品,对眼睛及皮肤具有刺激性,进入环境后会对动植物生长发育产生不良影响,且对生物体有致癌、致畸、致突变风险,被公认为是一种有毒难降解的有机化合物。近年来,喹啉和异喹啉在一些染料产品中已被检出,由于其本身所具有的毒害性和难降解性,使之在染料产品后续应用时会部分或全部转移到纺织品中,从而危害消费者健康。ECHA已将喹啉归类为CMR物质(致癌、致突变或致生殖毒性),并在“纺织品中的CMR物质”主题下对喹啉进行了讨论。2018年,OEKO-TEX®协会发布了STANDARD 100产品认证的检测标准,提出了纺织产品中喹啉的检测要求[2]。2019年1月最新发布的STANDARD 100则进一步明确规定其限量值为50 mg/kg。目前,已有报道对喹啉或异喹啉进行检测,如季浩等采用气相色谱法测定染料产品中的喹啉[3],巴德彪等采用气相色谱法测定化工品中的异喹啉[4],吴银菊等采用气相色谱-质谱联用法测定卷烟烟气中的喹啉[5],赵建宏等采用液相色谱法测定电合成反应液中的喹啉和喹啉酸[6],杨鹏等采用液相色谱-Q TOF MS法测定了白屈菜中的异喹啉类生物碱[7]。另外,徐龙鹤等采用气相色谱/氢火焰离子化检测器(FID)即GB/T 31531-2015[8]测定了染料及染整助剂中喹啉。但以上方法仅针对喹啉或异喹啉中的一种进行测定,并不适用于两者同时测定。因此,本文建立了GC-MS测定纺织品中喹啉和异喹啉的方法,弥补了现有测定方法的技术空白,提高了方法的检测灵敏度和效率,相关方法尚未见报道。
1 实验部分
1.1 仪器、试剂与材料
Agilent 7890A-5975C气相色谱-质谱联用仪(美国Agilent公司);R215旋转蒸发仪(瑞士Buchi公司);超声波发生器(昆山市超声仪器有限公司)。0.22 μm针式滤膜过滤头(津腾实验室设备有限公司)
喹啉、异喹啉标准品(纯度≥98%,德国Dr.Ehrenstorfer公司)。乙酸乙酯(色谱纯,美国Fisher公司),经5A分子筛脱水处理。喹啉、异喹啉标准品均用乙酸乙酯配成1 000 mg/L标准储备液,于4 ℃下储存,使用时用乙酸乙酯逐级稀释至所需浓度。
1.2 样品前处理
取5.0~10.0 g代表性纺织品样品,剪成5 mm×5 mm以下的碎片,混匀。精密称取1.0 g(精确至1 mg)试样于样品瓶中,加入20 mL乙酸乙酯,超声提取20 min,再将提取液转移至鸡心瓶中,残渣用20 mL乙酸乙酯重复超声提取1次,合并提取液于鸡心瓶中。将鸡心瓶置于旋转蒸发仪上,于40 ℃水浴中缓慢浓缩至近干,再向其中加入1 mL乙酸乙酯,振荡,混匀。过0.22 μm滤膜,待测。
1.3 色谱-质谱条件
1.3.1 色谱条件色谱柱:HP-5MS毛细管柱(30 m×0.25 mm×0.25 μm);载气为高纯氦气(纯度为99.999%),载气流速1.0 mL/min;升温程序:初始柱温50 ℃,以20 ℃/min升至180 ℃,再以25 ℃/min升至280 ℃,保持4 min;进样口温度280 ℃,不分流进样,进样量1 μL。
1.3.2 质谱条件溶剂延迟4 min;电子轰击能量70 eV;离子源温度230 ℃;四极杆温度150 ℃;接口温度280 ℃;扫描方式:SCAN、SIM,扫描范围:40~200 amu;定性离子:m/z129、102、76、51。
2 结果与讨论
2.1 喹啉与异喹啉的GC-MS图谱特征
由于喹啉和异喹啉属同分异构体,因此实现有效分离以及准确定性定量是重点解决问题。程序升温色谱分析中,升温速率是影响保留时间和分离度的主要参数,两个相邻难分离组分的分离度与升温速率成反比。本文在载气流速为1.0 mL/min下,考察了不同升温程序对喹啉和异喹啉分离的影响,结果表明,在“1.3.1”所述升温条件下,两者能够实现较好的分离,且保留时间均在8 min内。通过对混合标准溶液进行质谱全扫描得到总离子流图,选择目标分析物特征碎片离子作为定性和定量的目标监测离子。从两者质谱图看,喹啉和异喹啉的碎片离子及其丰度比基本相同。因此,将喹啉和异喹啉的碎片离子m/z129、102、76、51作为定性离子,其中分子离子峰m/z129作为定量离子(图1)。
2.2 前处理条件优化
2.2.1 提取溶剂的选择由于喹啉和异喹啉在有机溶剂中溶解性高于水,因此选择甲醇、丙酮、乙腈、乙酸乙酯、二氯甲烷、苯、甲苯7种有机溶剂对自制阳性纺织品样品(选用棉白色贴衬织物作为阴性基质样品,取不含喹啉和异喹啉的直接染料,向其中加入一定量的喹啉和异喹啉标准物质,仿照纺织品染色工艺流程,将布料在染料中充分浸泡上色,晾干后即成)进行测定,考察不同提取溶剂对纺织品中喹啉和异喹啉的提取效果。结果显示,苯系溶剂(苯、甲苯)和乙酸乙酯提取效果均较好,优于其他4种溶剂。但苯系溶剂的毒性较大,且提取液中共溶杂质较多,易对定量结果造成干扰。因此,本实验选择乙酸乙酯为提取溶剂。

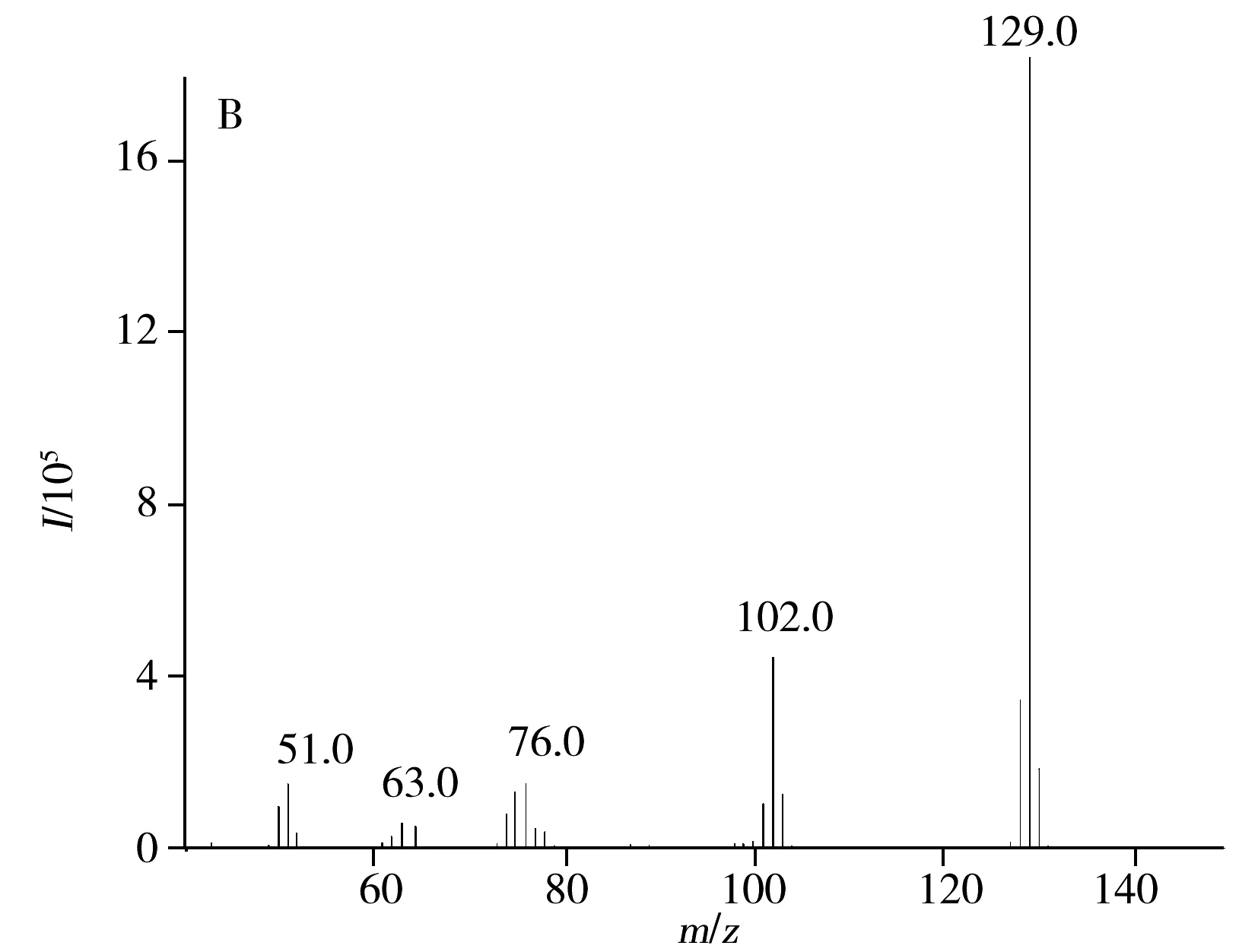
图1 喹啉、异喹啉的总离子流色谱图(A)和质谱图(B)
2.2.2 提取方式的选择常用的提取方式有超声辅助提取法、索氏提取法、固液振荡萃取、微波萃取、加速溶剂萃取等。其中索氏提取效率较高,但过程繁琐,提取时间长,试剂消耗量大。加速溶剂萃取设备昂贵,普及率不高。基于方法标准的普及型和可操作性,本研究比较了固液振摇提取和超声水浴提取的提取效果,结果发现,2种提取方式所得结果无显著差异,但固液振荡提取平行性稍差。因此,本研究选择超声提取方式进行样品前处理。
2.2.3 提取时间与温度的选择实验考察了常温条件下超声萃取时间(10、20、30、40 min)对提取效果的影响。结果显示,超声提取10 min时的回收率偏低,而超声20 min和30 min的回收率差别不大,超声40 min的回收率最高。但操作时间过长,易导致提取溶剂温度升高,方法精密度偏差较大。故本方法最终采用超声提取20 min。另外,实验还比较了不同提取温度(常温、40 ℃、50 ℃)超声提取20 min的提取效率。结果发现提取温度对提取效率的影响不大,但较高的提取温度更易导致溶剂挥发,从而影响提取效果,因此,实验选择常温下进行超声提取。
2.2.4 提取次数的选择取阳性样品,加入20 mL乙酸乙酯,按“1.2”所述实验条件提取3次,分别定容后上机测试。结果显示,对于阳性样品,使用20 mL乙酸乙酯提取时,第1次即可达到95%的提取率;第2次提取后,回收率可达98%左右,第3次提取回收率几乎无变化。因此,方法选择使用乙酸乙酯提取2次。
2.3 线性范围与定量下限
分别配制喹啉和异喹啉质量浓度为0.05、0.5、1.0、5.0、10.0 mg/L的溶剂混合标准溶液和基质混合标准溶液,采用GC-MS分析。比较发现两者曲线斜率基本一致,表明基质效应影响较弱。因此采用溶剂标准溶液,以待测物的色谱峰面积(Y)对其质量浓度(X,mg/L)进行线性回归,绘制标准曲线。结果显示,喹啉和异喹啉在0.05~10.0 mg/L质量浓度范围内线性良好,线性方程分别为Y=1.66×105X-2 186(r2=0.999 9)和Y=1.42×105X-8 647(r2=0.999 8)。以10倍信噪比(S/N≥10)计算得喹啉和异喹啉的定量下限均为0.05 mg/kg。
2.4 回收率及相对标准偏差
以不含目标化合物的混纺织物为空白基质,分别添加低(0.05 mg/L)、中(0.5 mg/L)、高(5.0 mg/L)3个浓度水平的喹啉和异喹啉混合标准溶液,每个浓度平行6次,在优化条件下测定,计算回收率。结果显示,喹啉和异喹啉在3个加标浓度下的加标回收率为82.0%~99.8%,相对标准偏差(RSD)为0.9%~3.8%,该方法完全满足痕量分析的要求。

图2 阳性样品色谱图
2.5 实际样品检测
采用本方法对87批纺织品中喹啉和异喹啉进行检测,均未检出喹啉和异喹啉。对自制阳性样品进行测定,测得喹啉和异喹啉的含量分别为4.25 mg/kg和4.17 mg/kg。图2为自制阳性样品中喹啉和异喹啉的GC-MS色谱图。
3 结 论
本文建立了纺织品中喹啉和异喹啉的GC-MS同时分析方法,可在8 min内实现喹啉和异喹啉的有效分离以及准确定性定量,方法的定量下限为0.05 mg/kg,远低于目前标准方法中10 mg/kg的检测要求,且方法操作简便,分析时间短,回收率和精密度较好,能够满足实际样品中喹啉和异喹啉化合物的测定要求。
