遵义地区莽椒细菌多样性及PICRUSt基因功能预测分析
2020-03-19崔梦君王玉荣葛东颖张振东刘欣郭壮
崔梦君,王玉荣,葛东颖,张振东,刘欣,郭壮
(湖北文理学院 食品科学技术学院,鄂西北传统发酵食品研究所,湖北 襄阳,441053)
作为我国华中和西南地区特色传统发酵食品之一,莽椒(又称莽海椒)的制作工艺相对粗放,通常将籼米和糯米按照一定的比例混合捣碎成粉,塞入切开去瓤的红辣椒中入坛密封发酵一个星期即可。莽椒的制作环境相对开放且加工过程中无杀菌等工艺操作,这使得原料、加工器具和环境中的微生物得以保留,莽椒的制作方式为厌氧方法,为乳酸菌等厌氧细菌的生长提供了良好的条件。由此可见,莽椒的品质形成与自然微生物的发酵密不可分,但是目前关于莽椒微生物多样性的研究报道尚少。
与莽椒制作工艺相同,鲊广椒亦是以籼米和辣椒为主要原料密封发酵而成,两者不同点在于鲊广椒制作工程中通常将辣椒切碎后与籼米混合发酵[1]。近年来,以Illumina MiSeq为代表的第二代高通量测序技术广泛的应用于传统发酵食品微生物多样性的解析中[2-3],在快速和准确解析发酵基质微生物多样性的同时,实现了多样本的平行比较[4-5]。采用该技术,王玉荣发现Lactobacillus为湖北省当阳地区鲊广椒中的优势细菌[6]。相同的发酵基质,略有差异的制作工艺,莽椒和鲊广椒中微生物群系是否亦存在差异是值得探讨的问题。
本研究从贵州省遵义地区采集了7 个莽椒样品,使用Illumina MiSeq测序技术对其细菌多样性进行了解析,同时从MG-RAST数据库下载了8 个采集自湖北省当阳地区鲊广椒样品的测序数据[6]进行比较研究,并采用PICRUSt和BugBase软件对两者细菌种群的基因功能和表型进行了预测。通过本研究的开展以期为后续莽椒品质改善和安全性提升提供一定的理论依据。
1 材料与方法
1.1 材料与试剂
莽椒样本采集自贵州省遵义市协台坝菜市场和松桃菜市场(106°94′E,27°70′N),共采集7 个,编号依次为MJ1~MJ7。
QIAGEN DNeasy mericon Food Kit DNA基因组提取试剂盒,德国QIAGEN公司;MRS培养基、石蕊牛乳培养基和LB培养基,青岛海博生物技术有限公司;5×TransStartTM FastPfu 缓冲液、FastPfu Fly DNA Polymerase和dNTPs Mix,北京全式金生物技术有限公司。
1.2 仪器与设备
DG250型厌氧工作站,英国Don Whitley公司;CR21N型高速离心机,日本日立金属株式会社;vetiri梯度基因扩增仪,美国AB公司;Illumina MiSeq高通量测序平台,美国Illumina公司;ND-2000C微量紫外分光光度计,美国Nano Drop公司;R920机架式服务器:美国Dell公司。
1.3 实验方法
1.3.1 乳酸菌的分离鉴定
使用无菌手术剪将莽椒样品剪碎,取10 g加入100 mL石蕊牛乳培养基中37 ℃培养36 h[7]。采用倍比稀释法将培养物稀释至10-3~10-6梯度后涂布于MRS培养基(含有1.0%(质量分数)CaCO3)中,37 ℃厌氧培养48 h[8]。从菌落数为30~300的平皿中挑选含有透明圈且形态不同的菌落纯化3 次后,将过氧化氢酶实验阴性、革兰氏染色阳性的菌株暂定为疑似乳酸菌菌株。参照文献[8]中方法,采用CTAB法对疑似乳酸菌菌株的DNA进行提取,并对其16S rRNA全序列进行扩增。对琼脂糖凝胶电泳检测合格后的扩增产物进行清洁,并将其连接到T载体后转化至Escherichiacolitop10 中,阳性克隆子寄往测序公司进行测序,反馈回的序列拼接后在NCBI数据库中进行比对并构建系统发育树。
1.3.2 宏基因组DNA提取和Illumina MiSeq测序
使用QIAGEN DNeasy mericon Food Kit DNA基因组提取试剂盒对莽椒样品进行宏基因组DNA提取,使用带有7个碱基核苷酸标签(barcode)的338F/806R引物,按照文献[9]中的PCR扩增体系和扩增条件对细菌16S rRNA V3~V4区进行PCR扩增,检测合格后的扩增产物使用Illumina MiSeq PE300高通量测序平台进行高通量测序。
1.3.3 序列质控和生物信息学分析
参照文献[10]中的方法对下机数据拼接后进行质控,将错配率≥0.2、引物碱基错配数≥2 bp或barcode碱基有错配的予以剔除,进而筛选得到高质量的序列集。以QIIME(v1.70)平台为依托[11],使用PyNAST软件对序列进行标准比对和对齐[12];选取100%和97%相似度进行两步UCLUST划分,构建分类操作单元矩阵(operational taxonomic units,OTU)[13];使用ChimeraSlayer软件剔除含有嵌合体序列的OTU[14];从OTU中选取一条代表性序列,使用GREENGENE、SILVA和RDP数据库进行同源性比对[15],进而在门、纲、目、科和属水平上获得各OTU的分类学地位;在使用FastTree软件绘制系统发育进化树的基础上[16],计算Chao1指数、发现物种数、Simpson指数和Shannon指数进而对菌群的多样性和丰度进行评价;使用UniFrac距离进行主坐标分析,进而完成遵义地区莽椒和当阳地区鲊广椒中细菌的β多样性分析。
1.3.4 核酸登录号
本研究中所有序列数据已提交至MG-RAST数据库,登录号为mgp90774。同时本研究下载湖北省当阳地区的鲊广椒序列数据纳入分析,该数据在MG-RAST数据库的登录号为mgp80896。
1.3.5 PICRUSt功能预测和BugBase表型预测
基于GREENGENE数据库对质控后的高质量序列数据进行UCLUST划分和注释,使用PICRUSt软件对莽椒和鲊广椒样品中微生物的基因功能进行预测[17],并依照蛋白质直系同源簇数据库进行功能注释[18]。将OTU矩阵与样品的分类信息上传至BugBase网站(https://bugbase.cs.umn.edu/)进行表型预测[19]。
1.3.6 多元统计学分析
使用Wilcoxon test对莽椒和鲊广椒样本的α多样性、菌群结构和功能类别进行显著性分析;使用冗余分析对造成莽椒差异的功能类别进行甄别;使用Spearman秩相关性分析法对样本中优势细菌属与功能类别之间的相关性进行计算,选取相关系数>0.6,且校正后P<0.05菌群和指标,采用Cytoscape软件进行相关性网络图的绘制。使用R软件进行分析和绘图;使用Canoco 4.5软件进行冗余分析和绘图;使用Origin 2017软件进行绘图。
2 结果与分析
2.1 莽椒中乳酸菌的分离鉴定
本研究从7 个莽椒样本中共分离出17 株疑似乳酸菌菌株;同时通过16S rRNA基因序列分析进一步将分离菌株鉴定到种水平,分离株与标准菌株的系统发育树如图1所示。
由图1可知,本研究分离出的17 株乳酸菌分别鉴定为Weissellahellenica(希腊魏斯氏菌,1 株)、Lactobacillusbrevis(短乳杆菌,1 株)、L.pentosus(戊糖乳杆菌,10 株)、L.plantarum(植物乳杆菌,2 株)、L.alimentarius(消化乳杆菌,1 株)和L.fermentum(发酵乳杆菌,2 株)。值得注意的是,17 株分离菌中有16 株为乳酸杆菌。
2.2 基于门和属水平的莽椒中细菌构成分析
本研究使用Illumina MiSeq高通量测序技术对莽椒中细菌的多样性进行了解析,共产生295 133 条高质量序列,平均每个样本产生42 162条。经100%和97%划分和去除嵌合体后共得到10 747个OTU。莽椒中优势门和属相对含量的方格图如图2所示。

图1 基于16S rRNA的17 株乳酸菌与标准菌株的系统发育树Fig.1 Phylogenetic tree of 17 strains of lactic acid bacteria and standard strains based on 16S rRNA
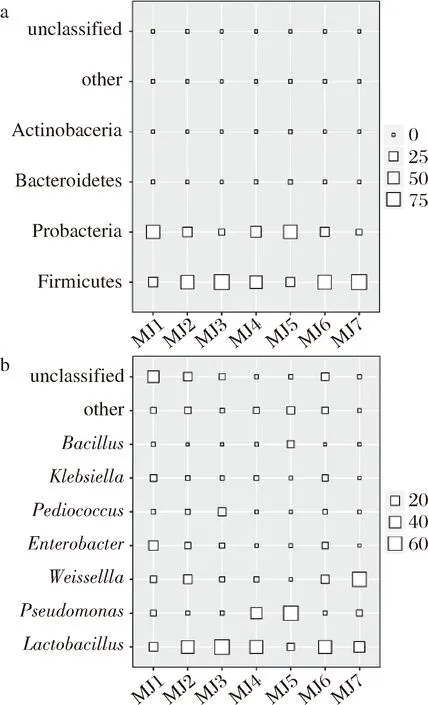
a-细菌门;b-细菌属图2 莽椒样品中主要细菌门和属相对含量的方格图Fig.2 The relative content of the main bacterial phylum and genus in the Mangjiao sample
由图2可知,10 747个OTU共注释到16个门,其中相对含量>0.1%的分别为硬壁菌门、变形菌门、Bacteroidetes(拟杆菌门)和Actinobacteria(放线菌门),其平均相对含量分别为63.15%、36.24%、0.30%和0.12%。由此可知,莽椒中的细菌主要隶属于硬壁菌门和变形菌门,而王玉荣的研究表明鲊广椒中的细菌亦主要隶属于硬壁菌门(62.36%)和变形菌门(25.97%)[6]。
由图2亦可知,10 747个OTU共注释到249个属,其中相对含量大于1.0%的分别为乳杆菌属(41.37%)、假单胞菌属(16.64%)、魏斯氏菌属(14.96%)、肠杆菌属(5.04%)、片球菌属(2.48%)、克雷伯氏菌属(2.40%)和芽孢杆菌属(1.33%),而王玉荣的研究表明鲊广椒中含量大于1.0%的细菌属也有7个,分别为乳杆菌属(77.33%)、魏斯氏菌属(2.24%)、片球菌属(2.18%)、葡萄球菌(1.27%)、Carnimonas(肉胞菌属,5.66%)、肠杆菌属(2.47%)和Prevotella(普氏菌属,1.05%)[6]。
2.3 莽椒与鲊广椒中细菌群落结构的比较
为了进一步甄别莽椒和鲊广椒中微生物类群的差异,本研究将王玉荣研究中涉及的序列从MG-RAST数据库中进行了下载[6],并将其与本研究获取的序列进行了归并,同时使用QIIME分析平台对两类样品的α和β多样性进行了解析,α多样性指数的比较箱型图如图3所示。

a-Cheol指数;b-发现物种数;c-Simpson指数;d-Shannon指数图3 莽椒和鲊广椒样品α多样性指数的比较箱型图Fig.3 Comparative boxplots of α diversity index of Mangjiao and Zhaguangjiao samples注:*代表P<0.05;ns代表差异不显著,P>0.05(下同)
由图3可知,莽椒中细菌在Chao1指数、发现物种数、Simpson指数和Shannon指数上均高于鲊广椒样本,经Wilcoxon test检验发现,Chao1指数和发现物种数在两者之间存在显著性差异(P<0.05)。Chao1指数和发现物种数常用于对样品中微生物的丰度进行评估,其值越大表明微生物丰度越高,由此可知,莽椒中细菌的丰度显著高于鲊广椒(P<0.05)。基于加权和非加权UniFrac距离的两类样品细菌群落结构的差异性分析如图4所示。
由图4-a可知,在基于加权UniFrac距离的主坐标分析图中,两类样品的空间分布存在交叠现象,经多元方差分析发现两组样品群落结构差异不显著(P>0.05);由图4-b可知,在基于非加权UniFrac距离的主坐标分析图中,两类样品呈现出明显的分离趋势,多元方差分析表明其群落结构差异极显著(P<0.001)。由此可见,莽椒和鲊广椒可能均各自含有一些含量较少但较为独特的细菌类群。经Wilcoxon test检验发现,变形菌门、假单胞菌属和魏斯氏菌属在莽椒样品中显著较高(P<0.05),而放线菌门和乳杆菌属显著较低(P<0.05)。
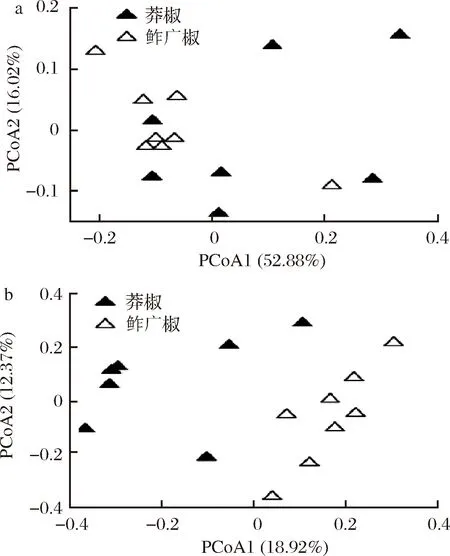
a-加权;b-非加权图4 基于加权和非加权的UniFrac距离的主坐标分析图Fig.4 Principal coordinate analysis diagram of UniFrac distance based on weighted and unweighted
本研究使用BugBase对莽椒与鲊广椒样品中细菌的表型进行了预测分析,结果如图5所示。
由图5可知,移动原件含量、生物膜形成、革兰氏阴性菌、致病潜力和氧化胁迫耐受在莽椒细菌中显著偏高(Wilcoxon test发现,P<0.05),而革兰氏阳性菌呈现出相反的趋势(P<0.05)。由此可见,莽椒与鲊广椒中的细菌在预测表型上存在着较大的差异。
2.4 PICRUSt功能预测
本研究进一步使用PICRUSt软件对莽椒与鲊广椒中细菌菌群的基因功能进行了预测。在预测基因功能的同时,参考蛋白质直系同源簇数据库(clusters of orthologous groups of proteins,COG)对预测的功能类别进行了注释。莽椒与鲊广椒样本中细菌功能类别的比较分析如图6所示。

a-革兰阴性菌;b-革兰氏阳性菌;c-致病潜力;d-厌氧;e-好氧;f-兼性厌氧;g-氧化胁迫耐受;h-生物膜形成;i-移动原件含量图5 莽椒与鲊广椒样品中细菌表型结果的比较分析Fig.5 Comparative analysis of bacterial phenotypic results in Mangjiao and Zhaguangjiao Samples

a-功能类别热图;b-功能类别冗余分析;c-显著性差异功能类别;A-RNA的加工与修饰;B-染色质结构与动力学;C-能量生产和转换;D-细胞周期控制、细胞分裂、染色体分割;E-氨基酸转运与代谢;F-核苷转运与代谢;G-碳水化合物运输和代谢;H-辅酶转运与代谢;I-脂质转运与代谢;J-翻译、核糖体结构与生物发生;K-转录;L-复制-重组和修复;M-细胞壁/膜/包膜生物发生;N-细胞运动;O- 翻译后修饰,蛋白质周转,伴侣;P-无机离子运输与代谢;Q-次生代谢产物的合成-转运和分解代谢;R-一般功能预测;S-未知功能;T,信号转导机制;U-细胞内运输,分泌和囊泡运输;V-防御机制;W-真核细胞的细胞外结构;Y-核外结构;Z-细胞骨架(下同)图6 莽椒与鲊广椒中细菌功能类别的比较分析Fig.6 Comparative analysis of bacterial functional genes in mangjiao and zhaguangjiao samples
本研究从莽椒和鲊广椒细菌中共注释到4 792个COG,这些COG分别隶属于23 个功能类别。由图6-a可知,碳水化合物的运输与代谢、氨基酸的转运与代谢和转录在两类样品中均高表达,而细胞运动、RNA的加工与修饰、染色质结构与动力学和细胞骨架的表达则相对较低。由图6-b可知,通过冗余分析发现有12 个功能类别对莽椒和鲊广椒细菌多样性的区分有影响。由6-c所示,3 个功能类别在莽椒和鲊广椒样品中存在显著性差异(P<0.05),莽椒样品中细菌的细胞内运输较强,而鲊广椒中细菌的细胞周期控制、细胞分裂、染色体分割和翻译、核糖体结构与生物发生的表达较强。两类样品中主要菌属(平均相对含量大于0.1%)与功能类别之间的相关性网络图如图7所示。

图7 主要菌属与功能类别之间的相关性网络图Fig.7 Correlation network diagram between dominant bacteria and functional classes注:实线表示显著正相关,虚线表示显著负相关;连线的粗细表示相关性的大小,线越粗相关性也大,反之越小
由图7可知,乳杆菌属、Salmonella(沙门氏菌属)、肠杆菌属、Lactococcus(乳球菌属)和克雷伯菌属与12 个功能类别之间具有显著相关关系(P<0.05),其中这12 个功能类别主要隶属于碳水化合物代谢、核酸代谢和蛋白质代谢相关的功能类别。值得注意的是,除乳杆菌属与细胞运动、胞内运输和信号传导呈显著负相关外(P<0.05),其他菌属与功能类别之间呈显著正相关(P<0.05)。由此可见,上述菌属在莽椒品质形成方面发挥着关键作用。
3 讨论
在现代食品加工产业中,乳酸菌是生产发酵乳、泡菜和益生菌制剂等常用的菌种之一,具有应用范围广、产品种类多和市场产值大的特点[20]。我国学者围绕乳酸菌菌株的挖掘、收集、高密度发酵、功效评价和产业化应用开展了大量卓有成效的研究,极大地促进了我国乳酸菌及相关食品产业的发展。目前我国开展乳酸菌研究的科研院所和高校多集中在东北三省、内蒙古自治区、新疆维吾尔自治区、北京市、广东省、四川省和江浙沪地区,而位于我国华中地区的湖北省及西南地区的贵州省开展乳酸菌相关研究的较少,且目前产业化生产中使用的乳酸菌菌株多来源自传统发酵乳制品、肠道和发酵蔬菜制品。以莽椒和鲊广椒为代表的特色发酵食品在我国华中及西南地区较为流行,其发酵基质和发酵条件与泡菜和乳制品均存在较大差异,通常以大米、玉米和辣椒为主要原料固态厌氧发酵而成,因而其中蕴含的乳酸菌资源可能较为独特和多样[21]。本研究从遵义地区莽椒中分离的绝大部分乳酸菌为戊糖乳杆菌,虽其在泡菜[22]和发酵乳[23]中亦有报道,但均为非优势乳酸菌。通过采用单分子实时测序技术,王玉荣等发现湖北省当阳地区鲊广椒中的乳酸菌主要为破布子乳杆菌(L.pobuzihii)、食品乳杆菌(L.alimentarius)和费斯莫尔德乳杆菌(L.versmoldensis),而这些细菌种在其他发酵食品中报道亦较少[24]。此外,ZHANG等亦从湖北省当阳地区的鲊广椒中分离到1株乳酸菌新物种,并将其命名为鲊广椒乳杆菌(L.zhachiliisp. nov)[25]。由此可见,我国华中及西南地区以莽椒和鲊广椒为代表的淀粉基质发酵食品中含有丰富且较为独特的乳酸菌群系,因而在对发酵食品微生物多样性进行解析的基础上进一步进行乳酸菌菌株收集,对后续乳酸菌遗传多样性研究和产业化推动具有积极的意义。
MiSeq测序结果显示,乳杆菌属和魏斯氏菌属是莽椒样品中的优势菌属,分别占到全部细菌含量的41.37%和14.96%。乳杆菌在莽椒的发酵过程中可以将糖类物质转化为乳酸,同时伴随着乙醇、乙酸和NO2等各种副产物的生成,不仅使莽椒形成了特有的风味,对杂菌和有害菌亦有一定的抑制作用。作为异型发酵乳酸菌,魏斯氏菌的主要代谢产物为乳酸和乙酸,有研究表明其可产生寡糖和胞外多糖(主要是葡聚糖),而这些物质可存在于发酵制品中为人类所利用[26]。本研究亦发现莽椒中存在大量的条件致病菌,肠杆菌属和克雷伯氏菌属细菌的占比达7.44%,通过基因功能预测亦发现莽椒中细菌具有较强的生物膜形成和致病潜力,究其原因在于莽椒的制作环境较为开放,发酵过程中极易受到家畜粪便和污水的污染。由此可见,在改善加工环境的同时,积极筛选具有优良发酵特性的乳酸菌进行莽椒的纯种发酵,对推动莽椒的产业化进程及提升食品安全性具有积极的意义[27]。
通过基因功能预测发现,莽椒和鲊广椒中细菌的碳水化合物运输与代谢功能均较强,究其原因可能在于两者制作工艺较为相似,均以大米、玉米和辣椒为主要原料。虽然莽椒和鲊广椒制作均为厌氧发酵,但基因预测发现莽椒和鲊广椒中细菌均含有较多的兼性厌氧菌且其氧化胁迫耐受能力亦较强,导致这种现象的原因可能是居民开坛取食食材时破坏了罐内的厌氧环境,氧气的进入对细菌的多样性产生了影响。本研究发现遵义地区莽椒和当阳地区鲊广椒均各自含有一些含量较少但较为独特的细菌类群,生产环境和制作工艺对发酵食品的细菌多样性具有显著影响,近年来越来越多的研究表明,不同地区的发酵食品不仅微生物群系存在较大差异[28],同一乳酸菌种种内的遗传多样性亦存在很大不同[29]。由此可见,在后续研究中进一步扩大莽椒和鲊广椒的样本采集范围和数量,在采用宏基因组学策略对其微生物多样性进行揭示的基础上,使用多位点序列分型(multilocus sequence typing, MLST)和比较基因组学手段对乳酸菌分离株遗传多样性进行解析是极为必要的。
