二脲基桥联β-环糊精手性液相色谱键合相的制备与性能评价
2020-03-18双亚洲张天赐李来生
双亚洲, 王 惠, 张天赐, 李来生
(南昌大学化学学院, 江西 南昌 330031)
随着手性化合物在药品和生命科学中的广泛应用,由立体异构导致的对映体在活性、毒性、代谢和残留方面的手性问题日益突出[1-3],手性拆分早已引起人们的普遍关注。开发新型的高选择性手性分离材料[4-6],并建立快速高效的对映体分离分析方法,对保障药品、食品和环境安全具有重要的研究意义。
环糊精(cyclodextrins, CD)作为第二代超分子化合物,具有内疏水外亲水的独特性质,能与许多化合物形成包合物,具有很强的手性识别能力,已被广泛地用作高效液相色谱固定相和毛细管电泳添加剂[7-9]。通过环糊精端口的衍生化,可引入多种作用位点,从而进一步提升了该类固定相的手性分离能力[10-12]。但常用的全衍生化易造成端口拥堵,加之β-环糊精腔体较小(约0.65 nm),也使得其手性识别范围受到一定的限制[13]。制备桥联环糊精是提高手性选择性的新途径,有望弥补上述不足。
桥联环糊精(bridged cyclodextrins, BCD)是两个或以上环糊精单元经功能桥基连接形成的一个有机整体。已在人工模拟酶、药物载体、分子识别、不对称催化、传感材料、生物智能器件和环境污染物处理等众多领域得到了广泛的应用[14,15]。研究证实,桥联环糊精可显著增强天然环糊精的分子结合能力和选择性,表现出比单个天然环糊精更好的分子选择性结合能力,通常单个环糊精与底物的包结常数(Ks)一般小于105L/mol,而桥联环糊精与底物的包结常数可与生物酶相当,达Ks>1011L/mol[16]。Liu等[17]发现桥联基团还可以充当“假空腔”提供额外的相互作用,与多个环糊精腔体协同包合客体分子,从而进一步增强其空间识别能力,这在很大程度上弥补了单个环糊精腔体小的局限性。尽管桥联环糊精已经在许多领域得到了广泛应用,但其“协同作用”在手性分离中并未被有效利用。Chang等[18]和袁黎明等[19]分别报道了两种桥联环糊精键合相,发现其对一些芳香族化合物的位置异构体和氨基酸衍生物表现出比单环糊精固定相更好的选择性。本实验室的周仁丹等[20]也制备了一种乙二胺桥联环糊精固定相,并成功地快速拆分了14种β-受体阻滞剂。综上所述,新颖的桥联环糊精的合成、纯化及其手性固定相的开发有待更深入系统地研究。
本研究利用活泼的双功能试剂六亚甲基二异氰酸酯与含胺基环糊精间的加成反应,在室温下设计合成了一种二脲基桥联环糊精,将合成的桥联β-环糊精键合到硅胶基质上制备出一种新型的二脲基桥联β-环糊精固定相(UBCDP)。通过质谱、红外光谱、元素分析和热重等手段对合成的桥联环糊精和新固定相进行表征。以三唑类农药、黄烷酮类、丹磺酰氨基酸类药物为手性探针,进行了色谱性能的评价,并探究了流动相组成、pH以及温度对手性分离的影响,通过与单β-环糊精固定相(CDCSP)比对,探讨了UBCDP的分离机理。实验表明,CDCSP的对映体选择性明显优于UBCDP。
1 实验部分
1.1 仪器与试剂
ZQ4000/2695液相色谱-质谱联用仪(美国Waters公司); 5700型傅里叶红外光谱仪(美国Nicolet公司); Vorio EL Ⅲ型元素分析仪(德国Elementar公司); Diamond TG/DTA型同步热分析仪(美国Perkin Elmer公司); AW-60色谱装柱机(美国Haskel公司); Milli-Q超纯水(美国Millipore公司); BSA-224S型电子天平(精度0.000 1 g,德国Sartorious公司); TGL-16C高速离心机(上海安亭科学仪器厂); KQ-100E型数控超声波清洗器(江苏昆山超声仪器有限公司); DF-2集热式磁力搅拌器(金坛市鑫鑫实验仪器厂); ZKF-030型电热真空干燥箱(上海实验仪器总厂)。
三嵌段聚合物P123(PEO20、PPO70、PEO20,相对分子质量约5 800)、六亚甲基二异氰酸酯(HDMI,纯度≥98.0%)、异氰酸丙基三乙氧基硅烷(IPTS,纯度≥98.0%)和外消旋标准品(黄烷酮类、氨基酸类,纯度均≥98.5%)购于美国Sigma公司;三唑类外消旋标准品(纯度≥98.0%)购于上海农药研究院;1,3,5-三甲苯(TMB,分析纯)购于阿拉丁试剂公司;甲醇(MeOH)和乙腈(ACN)为HPLC级,购于美国Tedia公司;正硅酸乙酯(TEOS)、β-环糊精、对甲苯磺酰氯、冰醋酸(HAc)、三乙胺(TEA)、N,N-二甲基甲酰胺(DMF)、丙酮等其他均为分析纯,购于国药集团上海化学试剂有限公司,其中DMF经CaH2除水后密封备用。实验用水为超纯水(电阻率>18.2 MΩ·cm)。
1.2 UBCDP的制备
UBCDP的制备路线见图1。先采用二异氰酸酯合成桥联β-环糊精(见图1中的D),然后以异氰酸丙基硅氧烷为偶联剂,将桥联β-环糊精固载到有序硅胶SBA-15表面,制得UBCDP。
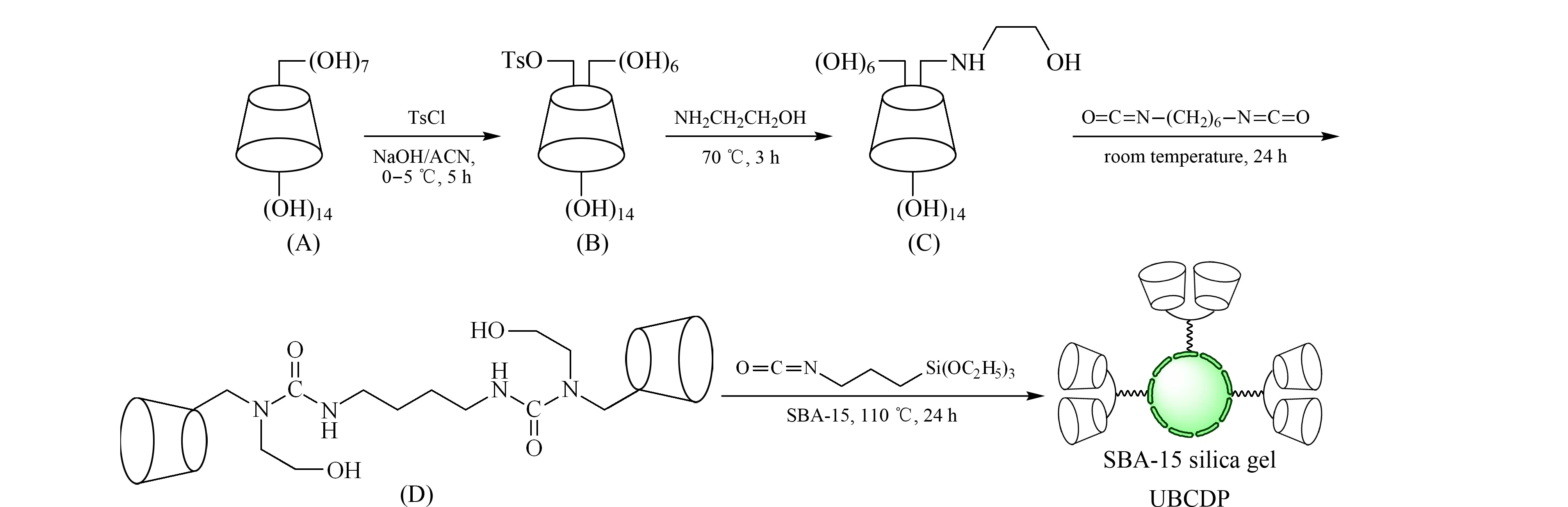
图1 UBCDP的合成路线
1.2.1SBA-15的制备
参考Zhao等[21]的合成方法,合成了有序介孔硅胶SBA-15,用作色谱键合基质。制备过程为:取5.6 g P123(模板)和12 g TEOS(硅源),溶于250 mL 2.0 mol/L HCl中,加入6.0 g TMB(扩孔剂),用适量的KCl调控其形貌,于38 ℃搅拌24 h,转入水热自压反应釜中,在110 ℃条件下水热反应静置晶化48 h,收集白色沉淀,烘干后在550 ℃高温下煅烧10 h除去模板,制得有序介孔硅胶SBA-15。其粒径大小约为3.2 μm,孔径大小约为24 nm,比表面积约为400 m2/g。后经稀盐酸浸泡过夜,以增加硅醇羟基数,并在160 ℃下除水来活化SBA-15表面的羟基,保存于干燥器中备用。
1.2.2二脲基桥联β-环糊精的合成
根据文献[20]合成单-6-去氧-(对-甲苯磺酰基)-β-环糊精(TsO-CD,见图1中的B)和单-6-去氧-6-羟基乙醇胺-β-环糊精(见图1中的C)。将60 g (52.9 mmol)β-环糊精(见图1中的A)加入300 mL蒸馏水中。边搅拌边滴加20 mL (8.25 mol/L) NaOH溶液至环糊精溶解。在0~5 ℃缓慢加入20.16 g (0.105 mol)对甲苯磺酰氯的乙腈(60 mL)溶液,在室温下搅拌反应5 h。滤除未反应的对甲苯磺酰氯,用盐酸调pH值至中性。于0 ℃冷藏过夜,过滤得粗品。用少量热水重结晶3次,干燥后得9.2 g TsO-CD,产率为13.5%。
将6.0 g (4.7 mmol)制备的TsO-CD(见图1中的B)加入50 mL乙醇胺中,于70 ℃反应3~4 h,冷却后倒入300 mL丙酮中,析出白色沉淀,将其溶于少量热水重结晶3次,于80 ℃真空干燥24 h,得4.32 g单-6-去氧-6-羟乙基胺-β-环糊精白色晶体,产率为78.6%。单-6-去氧-6-羟乙基胺-β-环糊精的质谱参数为:[M+H]+m/z=1 179.01(理论值为1 179.06)。
将上述合成的乙醇胺-β-环糊精(见图1中的C)溶于25 mL无水DMF中,然后加入0.31 g (1.84 mmol)六亚甲基二异氰酸酯,室温下反应24 h,反应结束后加入200 mL丙酮,析出白色沉淀,将过滤所得固体溶于少量热水,经C-25柱层析纯化。在65 ℃下真空干燥,得到1.52 g二脲基桥联β-环糊精(见图1中的D),产率为32.9%。二脲基桥联β-环糊精的质谱参数为:[M+H]+m/z=2 522.91(理论值为2 522.93)。
1.2.3UBCDP的制备
将上述合成的桥联β-环糊精在搅拌下溶于20 mL无水DMF中,慢慢滴加0.4 mL异氰酸丙基三乙氧基硅烷,滴加完后升温至80 ℃,反应2 h。然后加入4.5 g SBA-15,并补加适量无水DMF,于110 ℃反应24 h。过滤得粗品,用DMF、丙酮、甲醇、水反复洗涤,随后用丙酮索氏提取16 h清洗孔道,真空干燥后得到UBCDP。
称取1.0 g干燥的β-环糊精,溶于40 mL无水DMF中,加0.3 mL异氰酸丙基三乙氧基硅烷和3.0 g活化的SBA-15,其余的制备方法与UBCDP类似,得到单环糊精固定相CDCSP,用于对比研究。
1.3 色谱方法
1.3.1溶液配制
外消旋手性标准品的配制:将外消旋手性标准品用甲醇溶解,配制成质量浓度为100~200 mg/L的储备溶液,经0.22 μm滤膜过滤后放在4 ℃冰箱保存备用,使用前用甲醇或流动相稀释至适当浓度,并超声脱气处理。
流动相由水相和有机相组成。水相为水、1%(体积分数)甲酸或1%(体积分数)乙酸三乙胺(TEAA),其中1%(体积分数)TEAA是通过1%(体积分数)TEA水溶液用乙酸调节所需pH值制得。有机相为甲醇和乙腈。水相与有机相按一定比例混合,使用前用G4砂芯漏斗过滤,并超声脱气处理10 min。
1.3.2色谱柱的填装及柱效测定
采用匀浆填充法填装色谱柱。将上述自制的固定相超声分散在适量的丙酮中,转入匀浆罐。然后以甲醇作顶替剂,恒压(34.5 MPa)下将固定相匀填至一根不锈钢色谱柱(250 mm×4.6 mm)中,最后缓慢卸压。以萘为溶质,以甲醇-水(40∶60, v/v)为流动相,流速为0.8 mL/min,检测波长为254 nm,测得柱效为31 406plates/m。根据进样后的溶剂峰确定死时间(t0)为4.25 min。
1.3.3流动相组成与检测
流动相组成:水溶液、乙腈和甲醇,根据具体手性溶质的拆分需要按体积分数调整。根据被分析物和流动相的光谱性质,设定相应检测波长:黄烷酮类一般选择280 nm;丹磺酰氨基酸一般选择254 nm;三唑类农药一般选择220 nm(灭菌唑260 nm)。柱温为25 ℃,流速为0.8 mL/min,进样量根据溶质的摩尔吸光系数调整为3~10 μL。
1.3.4色谱性能评价参数
采用下列3个参数评价色谱性能:
保留因子:k=(tR-t0)/t0,t0为死时间(单位min),tR为溶质的保留时间(单位min)。
选择性因子:α=k2/k1,k1和k2分别为前后被洗出对映体的保留因子。
分离度:Rs=1.18(tR2-tR1)/(Wh1+Wh2),tR1和tR2分别为前后两对映体的保留时间(单位min),Wh1和Wh2分别为前后两对对映体的半峰宽。
2 结果与讨论
2.1 桥联β-环糊精的合成
通常采用双功能试剂与环糊精端口的羟基反应合成桥联β-环糊精,由于环糊精端口羟基较多,反应过程中易发生交联和聚合。因此一般要先对环糊精端口的羟基进行选择性的活化(如制备氨基衍生化环糊精),然后与活泼性双功能试剂(主要包括二异氰酸酯、二酰氯、二羧酸和二酸酐等)反应,这样能保证每个端基与单个环糊精反应。本实验利用六亚甲基二异氰酸酯与含胺基环糊精间的加成反应,在室温下设计合成了一种二脲基桥联β-环糊精。首先向环糊精端口引入羟乙基胺基活性位点,然后利用衍生基团与活泼的六亚甲基二异氰酸酯加成形成稳定的脲键,脲键既是氢键给体,又是氢键受体,提供手性拆分所需的作用位点,加之己基链柔性较强,有利于配合两个环糊精腔体间的协同包结作用。这条合成路线具有步骤简单、条件温和、产率较高等特点。
另外,本文选取比表面较大、孔道有序的SBA-15作为桥联环糊精的载体,有利于提高键合量,有序的孔结构也可加快溶质传质。由于其整体结构规整,可有效地减少多路径,从而能降低涡流扩散,实现快速手性分析。
2.2 结构表征
2.2.1桥联β-环糊精的表征
将合成的桥联β-环糊精通过质谱和红外光谱进行结构表征,结果分别见图2和图3。图2中m/z2 522.93为桥联β-环糊精的[M+H]+峰,与理论值(m/z2 522.91)相符,m/z1 262.70为双电荷离子峰,即[M+2H]2+/2峰,与理论值(m/z1 261.97)相近。结合质谱分析,证明本试验成功地合成了二脲基桥联β-环糊精。
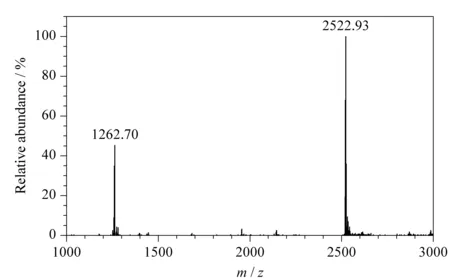
图2 二脲基桥联β-环糊精的质谱图

图3 二脲基桥联β-环糊精及UBCDP红外光谱图
2.2.2桥联β-环糊精固定相的表征
图3中上下分别为二脲基桥联β-环糊精和UBCDP的红外光谱图。图中3 391.15 cm-1处是O-H伸缩振动,2 941.66 cm-1处是桥基和环糊精上C-H伸缩振动,1 658.57cm-1处是脲基的C=O键伸缩振动,1 552.33 cm-1处是N-H键弯曲振动,1 098.75 cm-1附近的宽峰是环糊精上的C-O键和SBA-15的Si-O-Si键的重叠吸收峰。红外光谱分析表明合成的二脲基桥联β-环糊精被成功地键合到SBA-15硅胶上。
UBCDP的元素分析结果为:C为2.73%, H为0.27%, N为0.51%,以碳含量计算其键合量约为0.06 μmol/m2。

表1 二取代苯位置异构体分离结果
k1: the retention factor of the first peak;k2: the retention factor of the second peak;k3: the retention factor of the third peak;Rs1: the resolution of the first pair;Rs2: the resolution of the second pair; CSP: chiral stationary phase;m: meta;o: ortho;p: para; CDCSP:β-cyclodextrin-bonded chiral stationary phase; /: no separation.
CDCSP的元素分析结果为:C为3.82%, H为0.48%, N为0.13%,以碳含量计算其键合量约为0.18 μmol/m2。
热重分析:升温速度为10 ℃/min;记录温度为25~900 ℃。根据失重率(10%)计算得UBCDP的键合量约为0.10 μmol/m2,与按元素分析结果计算相近。
除光谱和元素分析结果外,该固定相具有良好的手性分离功能,也能进一步确证桥联β-环糊精被成功地键合到了硅胶上。
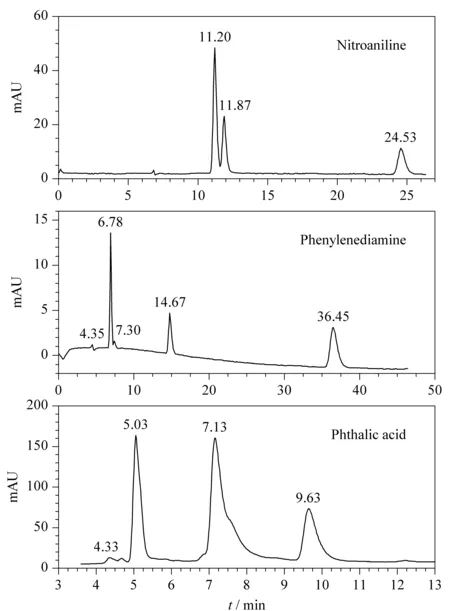
图4 UBCDP拆分二取代苯位置异构体色谱图
2.3 色谱性能评价
2.3.1二取代苯位置异构体的分离
采用甲醇-水(10/90, v/v)作流动相,利用自制的UBCDP对3种二取代苯位置异构体进行分离,并与CDCSP进行了对照研究,结果见表1和图4。可以看出,对硝基苯胺在最后出峰(m,o,p),这是由于线形的硝基苯胺更易进入环糊精腔体,包埋更深。而苯二胺和苯二酸的极性很强,邻位异构体易形成分子内氢键,大大降低其极性,与环糊精疏水性的腔体更加匹配,所以最后被洗脱。表明环糊精分离位置异构体的过程中,包结作用有重要贡献。
3种邻、间、对异构体的混合物在UBCDP上的保留时间明显更长,选择性和分离度都比CDCSP好。例如:UBCDP对中性、碱性(苯二胺)和酸性(苯二甲酸)异构体均有较强的分离能力,其中苯二甲酸的Rs高达23.68,苯二胺的Rs为3.68,而CDCSP却不能分离苯二胺位置异构体。可能是因为两个固定相对苯二胺的包结方式有所不同,桥联环糊精是通过两个腔体协同包结溶质,而单环糊精只靠一个腔体包结,这样桥联环糊精对m,p,o-苯二胺的识别作用更有效,故分离选择性好。
2.3.2黄烷酮在UBCDP上的分离
为评价桥联β-环糊精的手性色谱性能,本文采用不同结构类型的手性药物或农药作探针,包括酸性、碱性和两性的手性化合物,既可用于固定相色谱性能的评价,同时又为今后建立药物对映体的测定方法提供了理论依据。另外,通过相同探针在UBCDP和CDCSP上色谱行为的异同,探讨了UBCDP的色谱性能、特点及其分离机理。

图5 8种手性黄烷酮类化合物结构
黄烷酮(flavanones)及其衍生物大量存在于天然产物中,具有杀菌、抗炎、抗肿瘤等诸多药用价值,同时由于黄烷酮有众多可被取代位点,具备较大的结构修饰潜力,即作为手性药物合成的中间体。中草药的有效成分大多是含有天然的手性结构,因此进行手性分析有利于更深入地评价和制定药物质量标准。本文选取黄酮、2′-羟基黄烷酮、4′-羟基黄烷酮、6-羟基黄烷酮、6-甲氧基黄烷酮、柚皮甙、橙皮甙和儿茶素这8种黄烷酮类手性化合物(结构见图5),在反相色谱模式下进行手性拆分研究。通过与CDCSP的分离结果比对,探讨相关的拆分机理,试验结果和谱图分别见表2和图6。
鉴于该类化合物有酸性酚羟基,在反相色谱中拆分时使用含1%(体积分数)甲酸的流动相抑制其电离。从表2可看出,UBCDP对8种黄烷酮均有拆分能力,对映体Rs为1.06~4.35。其中2′-羟基黄烷酮和4′-羟基黄烷酮在30 min内Rs可达4.35和3.47。可是,CDCSP只能拆分其中的3种,且Rs(1.12~1.67)较低。尽管CDCSP的键合量(0.18 μmol/m2)比UBCDP(0.06 μmol/m2)高,但手性分离能力却较差。两种环糊精的端口均没有被衍生化,所以分离性能的差别应该与不同的包结方式有关。UBCDP通过两个腔体和柔性的桥基构成夹子形状[17],客体与桥联环糊精作用时并非进入某个腔体,而是整个客体被两个腔体和桥基协同包结,有利于对客体分子整体更精细的识别,包括R-和S-对映体空间结构上的差异[13]。单个β-环糊精腔体很小(约0.65 nm),至多能包萘环,而桥联环糊精不局限于腔体的大小,可与更大体积的客体形成协同包结配合物[14,15]。
由表2和图6可知,UBCDP可以拆分黄烷酮母体(Rs=1.47), 2′-羟基黄烷酮(Rs=4.35)和4′-羟基黄烷酮(Rs=3.47)。明显地,苯环上接有羟基有利于拆分,可能是黄烷酮与UBCDP形成包结物时,苯环上的羟基能与环糊精端口羟基、脲基存在氢键作用,从而产生良好的分离。从表2数据还可知,当黄烷酮上羟基或甲氧基的位置转移到苯并吡喃环上时,UBCDP对它们的分离度有所下降。例如6-羟基黄烷酮(Rs=1.14)、6-甲氧基黄烷酮(Rs=1.06)的Rs小于黄烷酮母体(Rs=1.47)和2′-羟基黄烷酮(Rs=4.35)。此时,CDCSP对它们没有分离能力。说明苯并吡喃环接有取代基(6-羟基或6-甲氧基)后体积变大,进入腔体较浅,单环糊精仅依赖单一腔体的包结难以有效识别它们的对映体。而桥联环糊精却有明显的优势,仍有一定的手性分离能力。
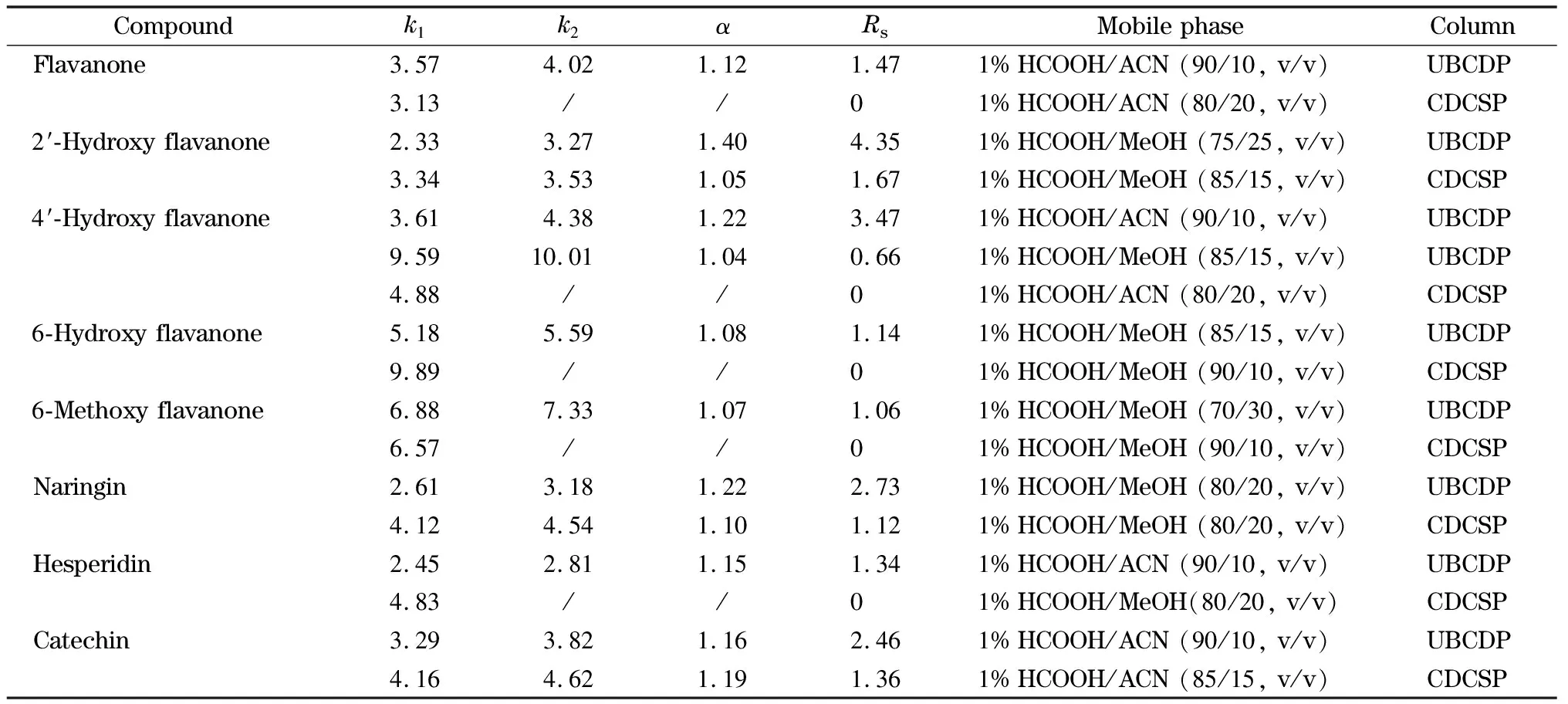
表2 8种黄烷酮类手性药物在UBCDP和CDCSP上的分离结果
α: selectivity factor; ACN: acetonitrile; MeOH: methanol.

图6 UBCDP拆分手性黄烷酮类色谱图
2′-羟基黄烷酮最容易被拆分,可能是2′-羟基与邻近的苯并吡喃环上的氧原子形成分子内氢键,使手性分子结构较刚性,有利于环糊精识别出R-和S-对映体的差别,从而实现良好的分离(Rs=4.35)。4′-羟基黄烷酮(Rs=3.47)是在含乙腈的流动相中拆分的,这可能是由于甲醇是氢键给体,而乙腈不是氢键给体,不会妨碍氢键作用的缘故。显然除包结作用外,氢键作用对手性分离也有重要贡献。例如含多个羟基的儿茶素、2′-羟基黄烷酮、柚皮甙和橙皮甙等Rs为1.34~4.35。特别是较大体积的橙皮甙,在单环糊精固定相上无法拆分,而UBCDP拆分橙皮甙的Rs分别能达到1.34。说明桥联β-环糊精两个空腔协同作用,在提高手性识别效率和拆分更大体积溶质方面存在优势[16]。
2.3.3丹磺酰氨基酸在UBCDP上的分离
氨基酸分子中同时有氨基和羧基两种官能团,是构成蛋白质的基本单位,许多生物大分子的构象和功能直接受氨基酸立体结构的影响,对生命科学具有重要意义。另外,氨基酸也是合成手性药物的重要中间体或原料。为了更好地拆分和检测这类高极性的两性化合物,通常需要衍生化,丹磺酰化的氨基酸是环糊精固定相的手性色谱性能评价的代表性探针。本文在反相色谱条件下,考察了UBCDP对8种常见丹磺酰氨基酸(结构见图7)的手性色谱性能和分离机理,分离结果见表3,色谱图见图8。

图7 8种丹磺酰化氨基酸的化学结构
反相模式下,在TEAA缓冲体系中,调节适当的pH值维持氨基酸分子上氨基和羧基的电离平衡,以便与固定相进行手性识别作用。如表3和图8所示,UBCDP对选定的丹磺酰氨基酸对映体均有拆分能力,其中对疏水性的丹磺酰化的亮氨酸(Dns-Leu)和苯丙氨酸(Dns-Phe)能达到基线分离,而在CDCSP柱上优化后也达不到基线分离(Rs<0.8)。说明疏水性氨基酸更易进入环糊精空腔而被包结,其中协同包结更紧密,对R-和S-对映体周围的环境识别更精细,所以分离效果更好,这与袁黎明等[19]的研究结果一致。从表3对其余6种丹磺酰氨基酸的分离也是桥联环糊精固定相占优势。
列举另一个例子,Dns-Asp属于典型的极性酸性氨基酸,含有两个可离子化的羧基,在反相色谱条件下与UBCDP作用力反而较强,需要25%(体积分数)甲醇和5%(体积分数)乙腈才能被洗脱,且能取得较高的分离度(Rs=1.32)。UBCDP和CDCSP两者的环糊精端口均没有进衍生化,后者键合量更高,推测应更有优势。可是实验结果相反,这说明协同包结、氢键等作用力可以提高桥联环糊精的手性分离能力[17]。
2.3.4三唑类农药在UBCDP上的分离
三唑类手性农药自1976年上市以来,品种一直在逐年增加,目前已经是全球种类最多、应用最广的一大类具有手性结构的杀菌剂和植物生长调节剂[22]。由于光学纯异构体合成很困难,大多数仍以外消旋体的形式销售使用。手性农药的不同对映体对靶标生物的生物活性、非靶标生物的生态毒性以及在动物和植物体内的富集行为往往存在显著的差异,美国已将三唑类农药列为潜在的手性致癌物[2]。本文在反相色谱模式下,选用常用的己唑醇、粉唑醇、灭菌唑、烯唑醇、三唑醇等9种三唑类杀菌剂作探针,进一步评价新的固定相拆分含三氮唑基的碱性农药的手性色谱性能。相关分离结果、条件及其代表性色谱图分别见表4和图9。
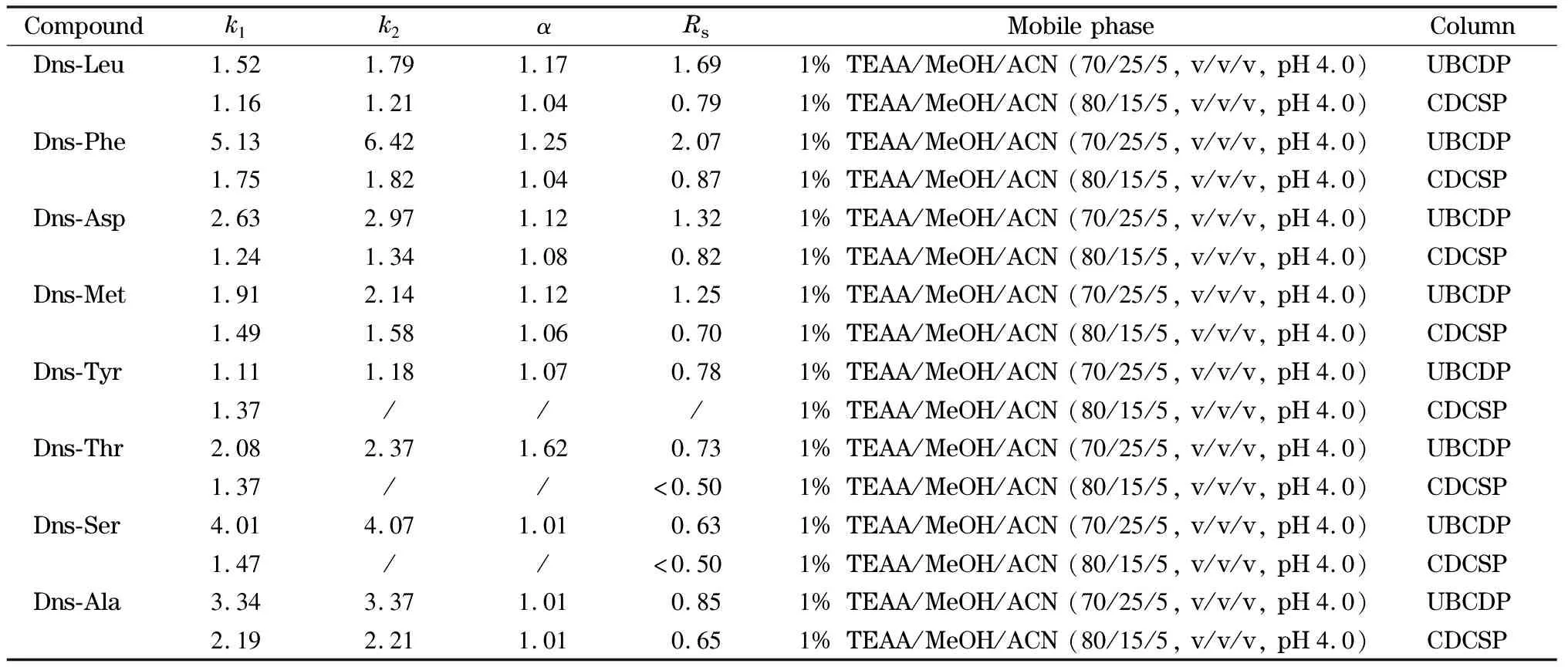
表3 8种丹磺酰氨基酸在UBCDP和CDCSP上的分离结果
TEAA: triethyl ammonium acetate.
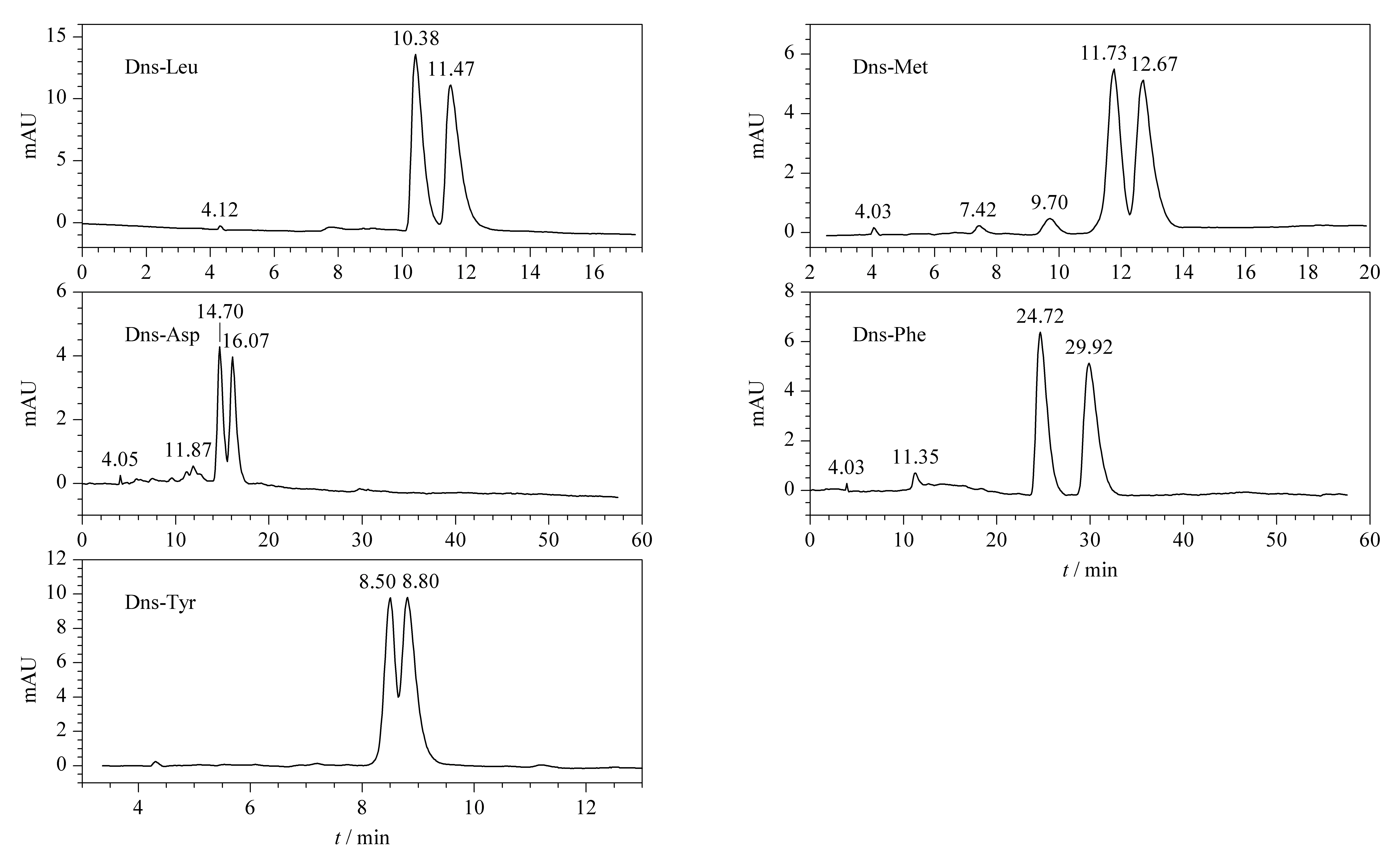
图8 UBCDP拆分手性丹磺酰氨基酸色谱图

表4 三唑类手性农药在UBCDP和CDCSP上的分离结果
a and b represent the enantio-resolution of two groups.
从上述图表中可以看出,采用简单的甲醇或乙腈和水组成的流动相,UBCDP对选定的三唑类农药均能进行较好的手性拆分,其中7种在25 min内达到或接近基线分离。UBCDP包结客体的芳基后,手性碳上的羟基与环糊精端口更接近,有利于提高手性识别效率。实验发现,MeOH或ACN含量对分离度的影响呈抛物线趋势,低浓度MeOH或ACN的流动相有利于溶质分子在固定相上保留,对拆分有利,但会造成溶质扩散,色谱峰展宽和拖尾,对映体选择性降低;当MeOH或ACN的含量在流动相中的比例较高时,溶质保留太短,还未与固定相作用完全就被洗脱,分离度也会下降。但甲醇和乙腈的比例会影响分离选择性,甲醇是质子溶剂,而乙腈不是,应该与氢键作用有关。因此,只有调整流动相中MeOH或ACN到合适的含量,才能取得较好的拆分结果。对于大多数三唑类手性农药溶质分子,实验发现,用MeOH/ACN/H2O的混合有机溶剂做流动相更有利于分离,但拆分抑霉唑(imazail)需要用TEAA,否则峰会展宽。
结合该类手性探针结构(见图10)和分离数据(见表4)可知:两固定相对手性碳上有羟基的己唑醇、粉唑醇、戊唑醇和烯唑醇对映体的分离较好,在23 min内Rs可达到1.52~1.58,实现了完全分离的目标。而对手性碳上不含羟基的三唑酮拆分十分困难,只有一点分离。三唑醇有两个手性碳,其中一个与羟基相连,可以被拆分,而另一个手性碳上没有羟基,与三唑酮类似,没有被拆分,故谱图上只能分出3个峰(见图9)。表明羟基作为氢键给体,可与固定相上的羟基或脲基形成氢键,这一作用对手性识别至关重要[18]。CDCSP对上述三唑类农药也展示一定的拆分能力,但达不到分离,最高的Rs为1.18。

图9 UBCDP和CDCSP拆分三唑类农药对映体的色谱图

图10 9种三唑类手性农药的化学结构
试验还发现,桥联环糊精固定相对灭菌唑的分离能力比CDCSP强。从图10可以看出,灭菌唑含有烯键,存在顺反异构体,其中顺式含量比反式低。顺式有2个对映体,反式有2个对映体,一共有4个对映体。从图9可看出,UBCDP能较好地拆分得到4个峰,Rs能达到1.42。可是优化后CDCSP只能分离其中顺式灭菌唑的2个对映体,而反式的2个对映体无法得到拆分。可能原因是桥联环糊精能通过协同包结和多重识别溶质,而普通环糊精只有单个腔体的包结作用,很难对灭菌唑进行较完整的分子识别。从表4可知,CDCSP也能拆分部分三唑类农药,但对烯效唑、灭菌唑、三唑醇和抑霉唑拆分效果差,甚至不能拆分。有关分离机理很复杂,有待多维NMR和高分辨质谱等技术做进一步的研究。
基于上述不同结构的手性探针的色谱性能表征,研究发现在端口没有被衍生的情况下,桥联环糊精固定相仍然展示了较出色的手性色谱性能,表明邻近腔体间,加上桥基互动可以协同包结溶质分子,协同包结作用和更多的氢键作用位点提高了桥联环糊精固定相的手性色谱性能。
3 结论
本文报道了一种新型UBCDP的制备方法。采用黄烷酮类、氨基酸类和三唑类等不同酸碱性的手性药物或农药探针,评价了新固定相在用途更广的反相色谱条件下的手性色谱性能。结果发现,无需端口衍生化,仅以简单甲醇或乙腈-水作流动相,成功地拆分了上述25种手性化合物,对4′-羟基黄烷酮的Rs高达3.45。与CDCSP比较,桥联环糊精具有较高的手性分离能力和较广泛的分离对象,主要得益于桥联双环糊精邻近腔体的协同包结作用和多羟基产生的丰富的氢键作用位点。桥联环糊精固定相在手性药物等质量监测和手性农药对映体残留量分析中存在广阔的应用前景。
