Screening and bioinformatics analysis of diabetic peripheral neuropathyrelated genes in women
2020-03-16YiZhangZhenQiuZhongYuanXia
Yi Zhang, Zhen Qiu, Zhong-Yuan Xia
Department of Anesthesiology, People's Hospital, Wuhan University, Wuhan, Hubei 430060
Keywords:diabetic peripheral neuropathy bioinformatics ESR1 gene CX3CR1 gene FGL2 gene
ABSTRACT Objective: To obtain the key genes and signal pathways of diabetic peripheral neuropathy (DPN) through bioinformatics analysis of related gene chips in the GEO database. Methods: The DPN-related gene chip was downloaded from the GEO database, and the differential genes (DEGs) between DPN female patients and the normal control group were analyzed and visualized using R language. According to the gene ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG), DEGs were annotated, their functions and related pathways were predicted, and a protein interaction network was constructed using the STRING database to screen for core genes. Results: The analysis chip GSE95849 obtained 4746 DEGs of which 2218 genes were up-regulated and 2528 genes were down-regulated. Among them, TFAP2C, ESR1, CX3CR1, and FGL2 are at the core site of protein interaction. Conclusions: Differential genes are mainly involved in the MAPK pathway. They participate in the pathogenesis of DPN through blood glucose homeostasis, inflammatory effects, and neuronal development, providing new ideas for the diagnosis and treatment of DPN.
1. Introduction
Diabetic peripheral neurophathy (DPN) is one of the common complications of diabetes. Up to 10% -20% of patients with type 2 diabetes have diabetic neuropathy when they are diagnosed with diabetes[1]. The main clinical manifestations of DPN are sensory and autonomic symptoms, including sensory disturbances, weakened tendon reflexes, numbness, and pain. The average time between patients and amputation is only two years. Their complex conditions, difficult diagnosis and treatment, and seriously affect the quality of life of patients[2-4]. The pathogenesis of DPN has not been fully elucidated, and its pathogenesis is currently thought to be related to genetic factors, metabolic disorders, oxidative stress, inflammatory response, nerve fiber loss and lifestyle[5-7]. The development of bioinformatics brings new ideas for the study of diseases. In this study, we searched the gene chip of DPN blood samples of GEO database for differential analysis, and screened differentially expressed genes (DEGs) of female patients with DPN and normal controls. Its function and signaling pathway, and the discovery of pathogenic core genes, are of great significance for the diagnosis and treatment of DPN.
2. Materials and Methods
2.1 Data set acquisition
The GEO database (https://www.ncbi.nlm.nih.gov/geo/) is a gene expression database created and maintained by the National Biotechnology Information Center (NCBI). The GEO database searches for the keyword "diabetic peripheral neurophathy" A data set, GSE95849, was obtained from the Affiliated Hospital of Ningbo University, using the Phalanx Human lncRNA One Array v1_mRNA platform. Blood samples were collected from 6 female DPN patients and 6 normal controls. Inclusion criteria for DPN patients: 1 confirmed type 2 diabetic patients; 2 hyposensory and positive neurosensory symptoms in the lower limbs (including tingling, burning or soreness, etc.); 3 distal sensation decreased, and ankle reflex was significantly reduced or missing; 4 abnormal sensory and motor nerve conduction. Patients with type 1 diabetes, cardiovascular disease, previous history of neurological disease, peripheral vascular occlusive disease, autoimmune disease, or any other disease that may cause peripheral neuropathy were excluded. The normal controls had the same gender, similar age and weight, and there was no statistical difference.
2.2 Standardized processing
After downloading the original matrix file, read the original data in R language and standardize the microarray data to make each group of data comparable and eliminate non-experimental differences between measurements. Non-experimental differences may result from sample preparation, hybridization processes, or hybrid signal processing. According to the annotation information of the chip, when a gene corresponds to multiple probes, average the probes or select one of the probes as the expression value.
2.3 Differential expression analysis
The limma package is currently the most widely used bioinformatics analysis package. It uses R language functions and the limma package to analyze the difference of the standardized chip expression profile, and uses Bayesian method to test multiple corrections. Differential genes were screened under the condition of log fold change> 1, and P <0.05 was considered statistically significant. And use R language to visualize the screening results, including volcanic maps and heat maps.
2.4 Function and Signal Pathway Analysis of DEGs
The DAVID database (http://david.abcc.ncifcrf.gov/) integrates biological data and analysis tools to provide systematic comprehensive biological function annotation information for largescale gene lists. Upload the selected DEGs to the DAVID database, and annotate the DEGs according to Gene ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG), including gene functions and biological pathways involved , Localization in cells, and signaling pathways involved. The screening conditions were P <0.05.
2.5 Interaction analysis of DEGs-encoded proteins
The STRING database (https://string-db.org/) is one of the most comprehensive protein databases. It currently stores 5090 species, 24'584'628 proteins, and a total of 3'123'056'667 protein interactions. Information. The DEGs were imported into the STRING website, and the protein interaction network analysis was constructed using Cytoscape software to predict the interactions between the proteins encoded by DEGs and screen out the key genes with the highest degree of connectivity to other genes.
3. Results
3.1 Standardized processing
Raw data may have missing values, genes correspond to multiple probes, etc. The chip expression profile is pre-processed and standardized using RMA method. The quality of the chip samples before preprocessing is shown in Figure 1, and the quality of the chip samples after preprocessing is shown in Figure 2.
Figure1. Sample quality box plot before pre-processing
Figure2. Sample quality box plot after pre-processing
3.2 DEGs screening results
There are 4746 differential genes in the data set GSE113439, of which 2218 genes are up-regulated and 2528 genes are downregulated. The specific distribution is shown in Figure 3. The ten genes with the largest differences are: ESR1, TFAP2C, CX3CR1, GIMAP4, MPEG1, SLCO4A1, FAM228A, C9orf64, FGL2, ZMYM6NB, as shown in Table 1. Among them, ESR1, TFAP2C, SLCO4A1, and FAM228A are up-regulated genes, and CX3CR1, GIMAP4, MPEG1, C9orf64, FGL2, and ZMYM6NB are downregulated genes. Fig. 4 is a heat map for selecting the ten genes.
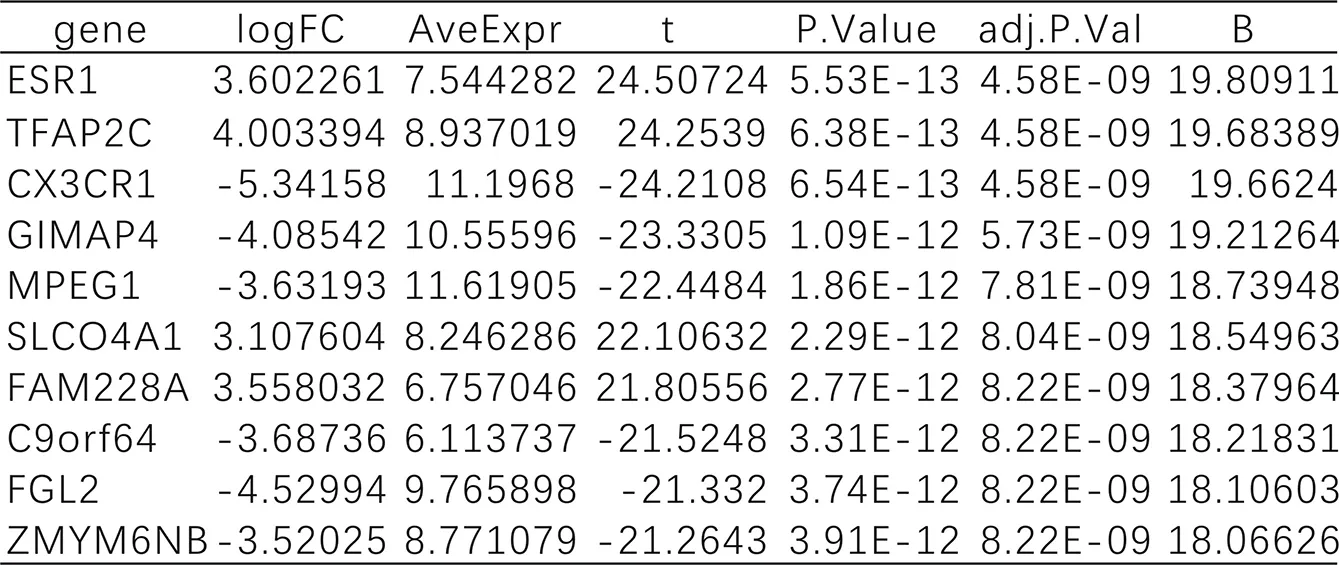
Table1.DEGs between two groups
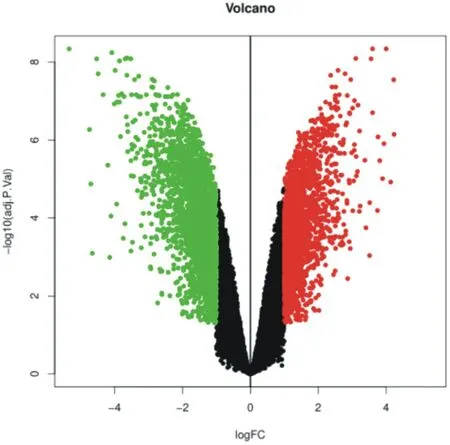
Figure3. Volcano plot of DEGs
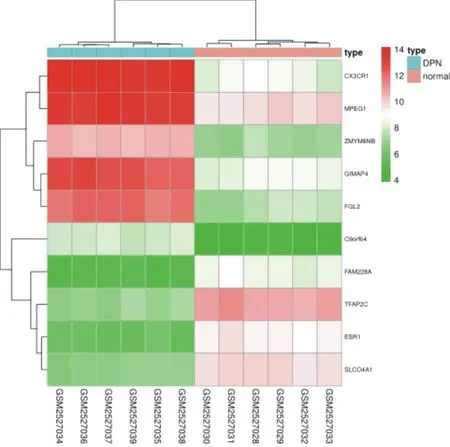
Figure4. Heatmap of DEGs
3.3 GO analysis and KEGG analysisGO
The enrichment analysis results showed that DEGs are mainly located in the nucleus, cytoplasm, and cytosol. The molecular functions are mainly protein binding, metal ion binding, and DNA binding. They mainly participate in DNA-based transcription and transcription regulation, and RNA polymerase II promoter Biological processes such as positive regulation of transcription are shown in Figure 5. The results of KEGG enrichment analysis showed that the differential genes were mainly involved in the MAPK signaling pathway, as shown in Figure 6.
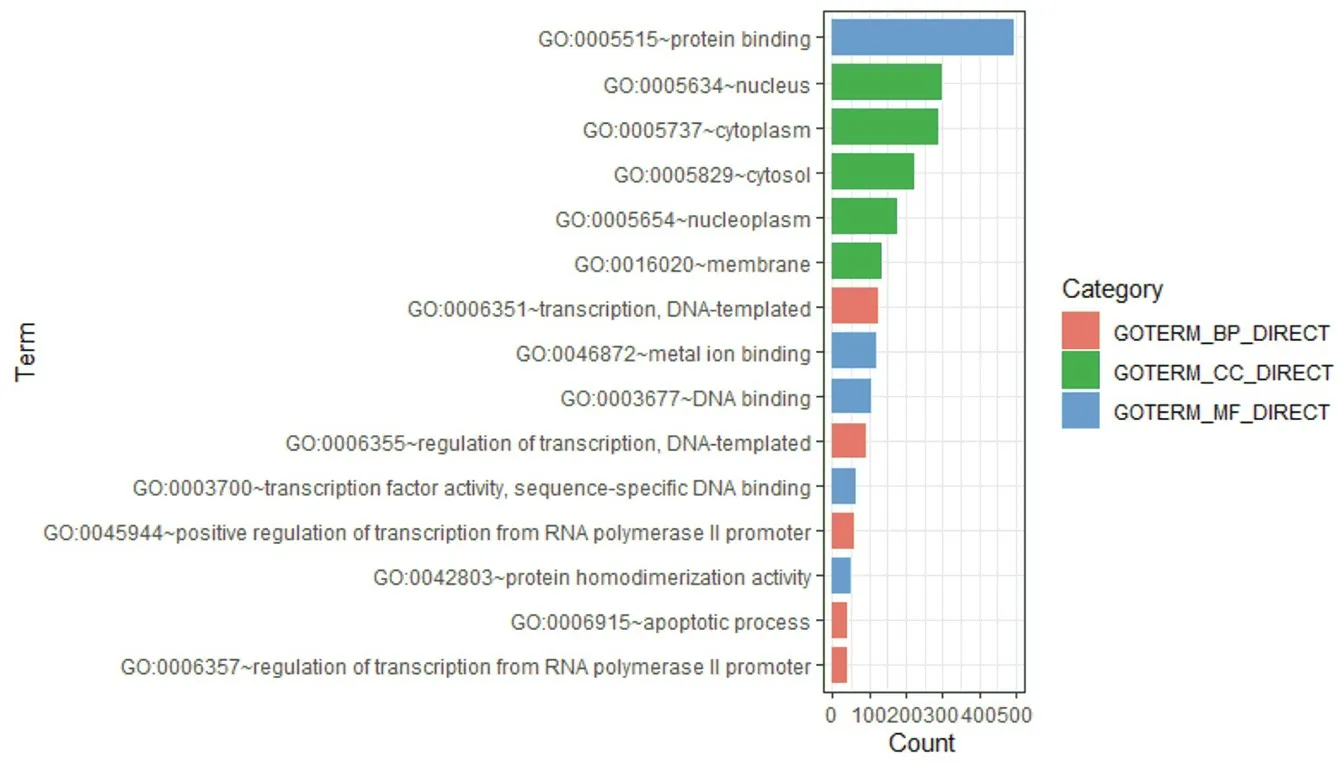
Figure5.biological function of DEGs
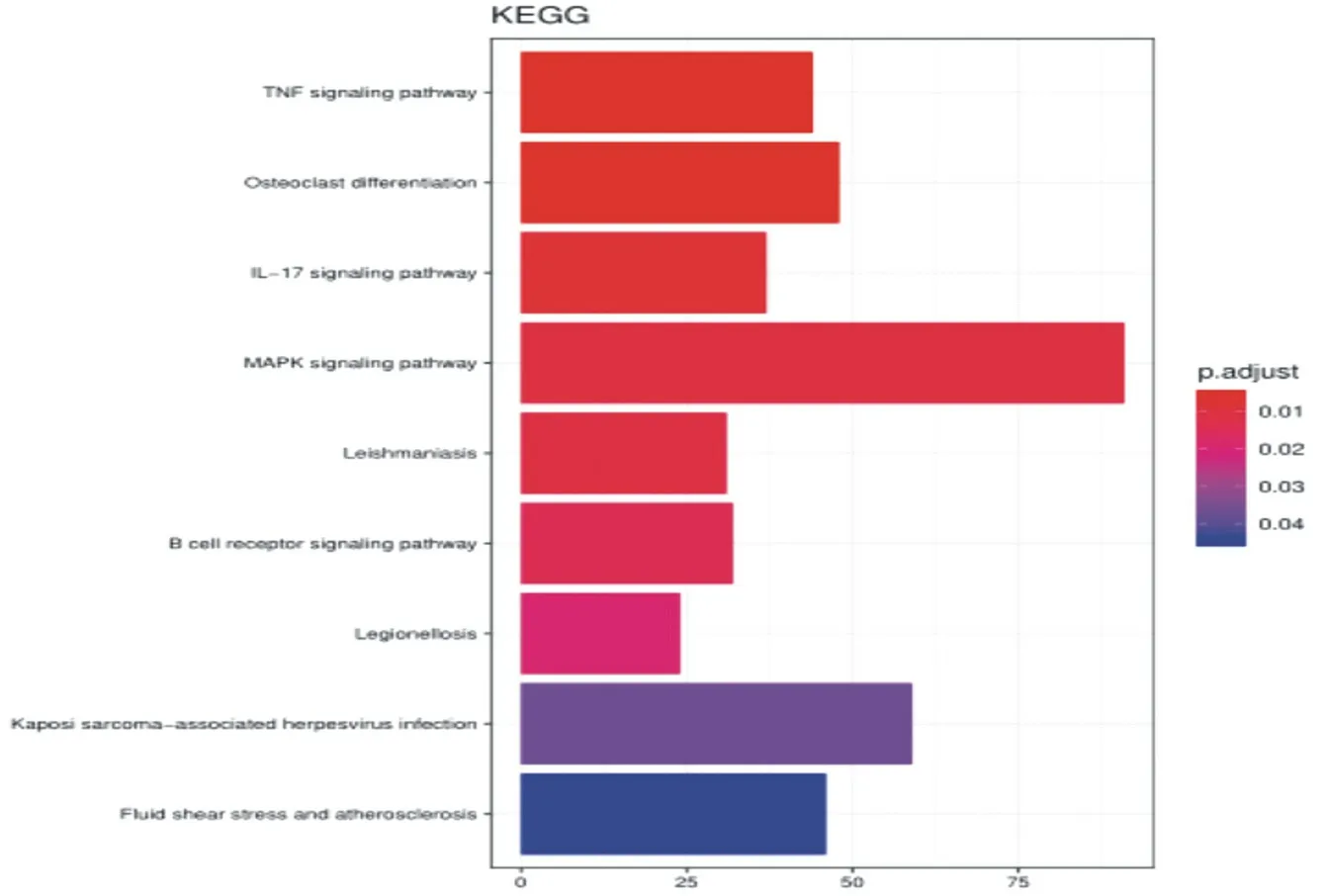
Figure6.pathways of DEGs
3.4 Protein interaction analysis
The STRING online tool and cytoscape software were used to analyze the protein interaction network of the 10 most different genes. The analysis results showed that TFAP2C, ESR1, CX3CR1, GIMAP4, FGL2, and MPEG1 proteins are more closely related to other proteins.
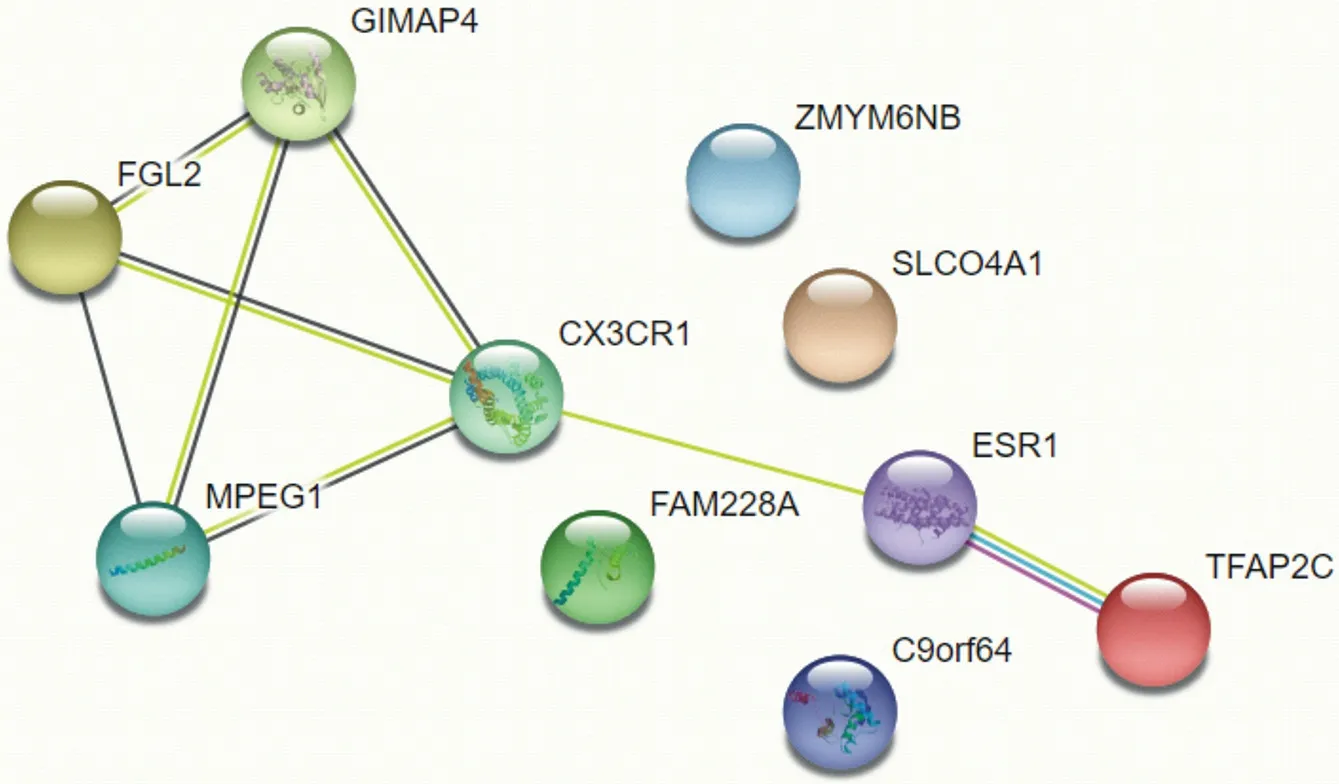
Figure7.protein interaction network
4. Discussion
Diabetes is a metabolic disorder syndrome characterized by chronic hyperglycemia caused by genetic factors, immune disorders, and insulin resistance. China became the world's largest diabetes country in 2010. It is estimated that by 2025, China's diabetes patients will reach 59.3 million [8]. Diabetic peripheral neuropathy is one of the most common complications of diabetes. Patients with diabetic peripheral neuropathy often experience sensory abnormalities such as hosiery-like sensations, numbness, burning pain, etc., which eventually cause lower extremity ulceration, gangrene, and even amputation, which greatly reduces the number of patients. Quality of life [9]. However, the onset of diabetic peripheral neuropathy is complicated, and there is no effective method for diagnosis. With the development of gene sequencing technology in recent years, a large amount of gene chip data for diabetes and diabetic complications have been uploaded to network databases. Data mining technology provides diagnostic and treatment new ideas for diabetes and diabetic complications including diabetic peripheral neuropathy, diabetic nephropathy, diabetic cardiomyopathy, etc [10-18].
In this study, a bioinformatics method was used to retrieve the diabetic peripheral neuropathy chip GSE95849 from the GEO database. Genetic data of 6 female patients with DPN and 6 normal controls were obtained and analyzed. A total of 4746 differential genes were found, of which 2218 were up-regulated and 2528 were down-regulated. These differential genes were subsequently analyzed by GO analysis, KEGG analysis, and protein interaction analysis. Among them, TFAP2C, ESR1, CX3CR1, and FGL2 were at the core of protein interactions. These genes may become diagnostic and therapeutic indicators of diabetic peripheral neuropathy.
TFAP2C transcription factor is involved in breast development, differentiation and tumorigenesis, and regulates the expression of ESR1 gene[19,20]. ESR1 (Estrogen receptor 1) gene, estrogen receptor 1, mediates the effect of estrogen on glucose homeostasis and plays an important role in pancreatic β-cell function and survival [21]. Elevated blood glucose is one of the basis of the pathogenesis of DPN. As a regulator of blood glucose homeostasis, estrogen has the ability to regulate glucose processing in muscle and adipose tissue, and is involved in the pathophysiology of obesity, insulin resistance and diabetes[22]. ESR1 is widely distributed in various organs and tissues related to glucose metabolism. It is a highly polymorphic gene that contains more than 1,600 single nucleotide polymorphisms (SNPs). Studies have shown that ESR1 variants PvuII, XbaI By altering the binding of its own transcription factors, it alters ESR1 gene expression, raises the levels of triglycerides, total cholesterol and LDL, and increases the risk of T2DM occurrence[23-25].
CX3CL1 is located on human chromosome 16 and is the only member of the CX3C (δ) subfamily of chemokines, acting through the sole receptor CX3CR1[26]. CX3CL1 is involved in many processes of human placental tissue, including inflammation and angiogenesis. When hypoxia or inflammation-induced secretion of inflammatory cytokines is strongly up-regulated, CX3CL1 may act as a key angiogenic factor locally[27]. Studies have shown that the deletion of CX3CR1 may impair microglial and macrophages' ability to promote the elimination of apoptotic cells or inhibit inflammation following phagocytosis, and accelerate the development of diabetic retinopathy in mice [28]. Similar to diabetic retinopathy, inflammatory effects are involved in the pathogenesis of DPN, and CX3CR1 is involved in the regulation of islet β-cell function and insulin secretion. Studies have found that CX3CR1 knockout mice show significant effects on glucose and GLP1 stimulated insulin secretion. Defects, and increased insulin secretion in mice and human islets after in vitro treatment with CX3C chemokine FKN[29]. Compared with the normal group in this study, CX3CR1 was significantly down-regulated in the DPN group, which means that the lack of CX3CR1 may play a leading role in the development of DPN through inflammatory responses and regulating insulin secretion[30,31].
Fibrinogen-like protein 2 (FGL2) is a new type of thrombinogenase that participates in microthrombosis and is also involved in apoptosis, angiogenesis and inflammatory responses. Studies have shown that it is involved in diabetic cardiomyopathy The occurrence and development of diabetic nephropathy[32,33]. In Zheng Zhenzhong et al.'S research on FGL2 and diabetic cardiomyopathy, it was found that FGL2 gene silencing inhibits cardiomyocyte apoptosis and improves cardiac function in diabetic rats induced by streptozotocin (STZ). The possible mechanisms include reducing p38 silk. Expression of mitogen-activated protein kinase (MAPK) [34]. In the development and function of neurons, the p38 / MAPK pathway phosphorylates other transcription factors, which in turn regulate the expression of genes. The products of these genes are involved in many aspects of neural development and function, including axonal growth, tree Synaptic pruning, synaptic function and plasticity[35]. This study confirmed that DPN patients have down-regulated FGL2. The KEGG study found that the MAPK pathway is down-regulated in DPN patients. It is speculated that down-regulated FGL2 and MAPK may be an important mechanism for the pathogenesis of PDN.
In short, through bioinformatics-based information mining technology, genetic difference analysis was performed between DPN patients and normal control groups, and 10 genes with the largest differences were screened out, and then 4 core genes were identified through the protein interaction network: TFAP2C, ESR1, CX3CR1 FGL2 is a potential DPN diagnostic gene. At the same time, these genes are mainly involved in MAPK and other pathways, and participate in the pathogenesis of DPN through blood glucose homeostasis, inflammatory effects, and neuronal development, providing new ideas for the prevention and treatment of DPN.
杂志排行

Journal of Hainan Medical College的其它文章
- Effect of Liraglutide Combined with Nephritis Rehabilitation Tablets for Type 2 Diabetic Nephropathy
- Study on TCM Syndrome Types and Syndromes of Knee Osteoarthritis Based on Modern Literature
- Study on the medication rule of Wang Xun in the treatment of dysentery based on Data Mining
- A Systematic Review and Meta-analysis for Duhuo Jisheng Decoction Combined with Manipulation in Treatment of Lumbar Disc Herniation
- Correlation analysis of osteonecrosis of the femoral head with blood lipid and coagulation indexes
- Patterns of Eye-Infiltrating Cells between Chronic and Acute Experimental Autoimmune Uveitis