黄河内蒙古段表层沉积物细菌多样性及群落结构类型
2020-03-16王晓丽其勒格尔
王晓丽,其勒格尔
1 内蒙古自治区环境化学重点实验室, 呼和浩特 010022 2 内蒙古师范大学化学与环境科学学院, 呼和浩特 010022
沉积物是水生态系统中氮、磷等物质循环的重要场所[1- 2]。沉积物微生物通过同化、异化等代谢过程来影响沉积物中氮、磷、碳等生源要素的矿化及生物地球化学循环过程,是水生态系统的重要组成部分[3],在维持水体环境稳定的过程中起着动植物无法取代的作用[4]。另外,微生物对其周围环境变化非常敏感,其生态系统中的微生物多样性及群落结构特征常可作为环境变化的指示因子[5-6]。因此,全面了解水生态系统中沉积物微生物群落组成及分布特征,对于管理和维护水生态环境具有深远的意义。随着分子生物学的发展,具有准确度高、通量大、成本低廉等优点的高通量测序技术在水体、沉积物、土壤微生物群落结构研究中得到广泛应用[7]。已有学者利用高通量测序技术对鄱阳湖典型湿地[8]、丹江口库区表层沉积物[9]、辽河口沉积物[10]微生物多样性和群落结构特征进行了研究。
黄河是中国北方的重要水系。黄河内蒙古段自宁夏入内蒙古境内后呈拱形走向,在榆树湾出境,全长约800多公里。内蒙古西部有五个沙漠,黄河内蒙古段河道泥沙淤积主要来源于内蒙古境内的乌兰布和沙漠和库布齐沙漠[11],每年借风力约有两亿吨沙尘刮入黄河流域,含沙量很大,在黄河所有河段中具有一定典型性和代表性。对黄河内蒙古段沉积物-水界面间的研究主要集中在生源要素磷[12- 14]、氮[15-16]在沉积物-水界面间的吸附解吸特征及其与沉积物理化性质间的关系,对黄河内蒙古段沉积物微生物多样性和群落结构类型研究较少。因此,本研究通过Illumina Miseq高通量测序技术分析黄河内蒙古段表层沉积物细菌多样性、丰度和群落结构类型,探讨细菌群落与环境因子间的关系,了解沉积物细菌对生源要素氮、磷转化的驱动作用,为其生物地球化学循环提供参考,并为黄河生态环境管理及微生物资源调控和利用提供科学依据。
1 材料与方法
1.1 研究区概况

图1 黄河内蒙古段采样点站位分布图Fig.1 Sketch map of sampling sites in Yellow River
黄河内蒙古段自宁夏入内蒙古境内后首先进入乌海市,呈几形走向,在榆树湾出境,全长约800多公里。黄河内蒙古流域贯穿内蒙古中部主要的工业、农业区,是沿岸城市居民生活用水的主要来源。流域内盆地是重要的粮食生产基地。同时人类的生产活动也日渐影响着黄河流域的水生生态系统。本实验所用沉积物样品分别从乌海市、临河、乌拉特前旗、包头市、托县和老牛湾6个地区采集,沉积物样品编号依次定为H1、H2、H3、H4、H5、H6。乌海市是典型的煤矿区域,受周围煤矿及洗煤厂的影响比较大;临河、乌拉特前旗是内蒙古典型的农业区,属于著名的河套平原区域;包头市是内蒙经济相对发达的工业城市,有包钢等大型企业;托县为普通小型县城;老牛湾是著名旅游景点,在夏季及秋季每年参观和游览的人较多。6个采样点分布图见图1。
1.2 样品采集与沉积物理化测定
采样时间为2016年7月份。在样品采集现场,用便携式水质监测仪现场测定沉积物的温度、氧化还原电位、电导率、溶解氧和pH。选用大小适宜的柱状采样器采集0—10 cm表层沉积物,每个采样点采集3个重复样,放入采集袋中,冰箱冷藏待测。上覆水水质指标见表1。
黄河表层沉积物样品分析指标包括阳离子交换量(Ion exchange capacitiy)、有机质(Total organic carbon)、总磷(Total phosphorus)、烧失量(Burning vector)。阳离子交换量参照EDTA-铵盐快速法测定,有机质采用重铬酸钾法来测定,总磷测定采用过硫酸钾氧化消解法测定。
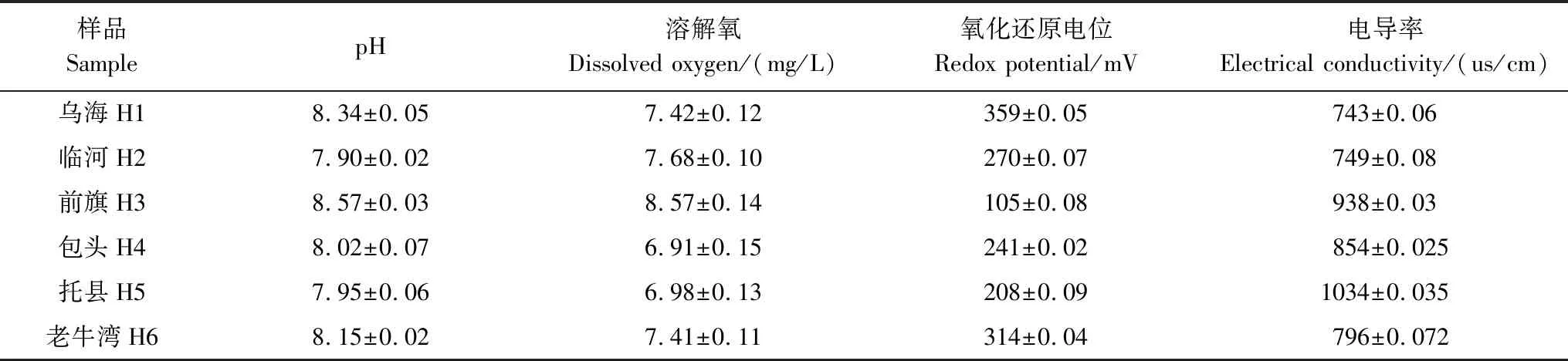
表1 上覆水水质指标
1.2 样品分析
1.2.1PCR扩增
提取的样品DNA先用琼脂糖凝胶电泳检测,确定检测合格之后再通过荧光定量PCR技术检测。Bact-F(5′-GTTAATACCTTTGCTCATTGA- 3′)和Bact-R(5′-ACCAGGGTATCTTAATCCTGTT- 3′)为本文选用的引物。其中,PCR反应体系包含2.5 μL 10 × PCR 缓冲液,1.6 μL dNTP (2.5 mmol/L,TaKaRa),1 μL 上下游引物(10 μmol/L), 0.125 μL rTaq DNA 聚合酶(5 U/μL),模版 DNA 2 μL,最终用去离子水补充至16.73 μL。反应条件设置为95 ℃ 预变性4 min,30个循环,95 ℃变性30 s、57 ℃退火30 s、72 ℃延伸30 s,最终72℃延伸10 min。最终要得到符合MiSeq高通量测序要求的目标DNA片段。
1.2.2Miseq平台高通量测序
16S 数据库测序是指对环境样品微生物的 16S rDNA 基因的PCR 扩增产物进行高通量测序后将测序数据与已有的 16S rDNA 数据库进行比对分析,对环境样品中的微生物群落多样性进行研究。其中PCR 反应的30 μL体系包含15 μL 高保真PCR混合剂(New Eng-land Biolabs),0. 2 μmol/L 的正反引物以及 2 μL的模版 DNA。PCR反应条件为 95 ℃预变性5 min,40个循环,57 ℃退火30 s,72 ℃延伸20 s,72 ℃最终延伸5 min。PCR产物通过琼脂糖凝胶电泳检测并确定合格后在Illumina MiSeq 平台进行更进一步测序。
1.2.3测序数据处理
扩增序列的处理主要包括截去条形码和引物序列。最终通过Flash1.2.7软件对序列进行拼接和用Qiime19.1.0等软件进行更加精确与严格的序列质量检测和筛选得到有效序列。其中碱基测序精确程度用Q值表示,然后采用UCLUST等方法进行OTU(operational taxonomic unit)聚类,并将把物种操作单元之间的相似性等于或大于97%的有效序列归为同一分类单元。基于OTU分析结果可以进行多种样品稀释性曲线分析并计算Chao1丰度指数、覆盖度(Coverage)和香农(Shannon)多样性指数。采用RDF Classifier贝叶斯算法对OTU代表序列进行分类学分析。
2 结果与分析
2.1 沉积物样品微生物多样性分析
群落生态学中,α多样性主要关注单样本的多样性分析,可以反映微生物群落中物种的数目,通过一系列统计学指数的分析来估计环境群落的物种丰度和多样性。6个采样点的沉积物样品所获得的多样性数据列于表2。由表2可知,6个采样点的沉积物样品ACE多样性指数在6758.63—12958.81之间,Chao1多样性指数范围在6079.61—11525.45内。ACE和Chao1多样性指数表明沉积物样品细菌丰度,其数值越大,样品细菌丰度就越高。6个样品细菌群落丰度大小为:H3 > H6 > H2 > H4 > H5 > H1。香农威纳多样性指数在9.46—10.57以内,其数值比较高,说明6个采样点都具有比较高的微生物多样性,其排序结果为:H6 > H2 > H5 > H4 > H3 > H1。与其他5个采样点相比,H1点的细菌群落丰度及微生物多样性明显都比较低。表中Coverage表示测序深度,从其值来看,除了H1和H5较低外,其余数值都比较高,高达96%,说明各样本文库的覆盖率比较全面,序列没有被测出的概率较低。该结果与本研究中稀释曲线预测结果一致。

表2 微生物多样性一览表

图2 黄河沉积物样品的主坐标分析 Fig.2 Principal coordinate analysis in sediments from Yellow River
黄河内蒙古段6种表层沉积物样品主坐标分析如图2所示。由图可见,第一主轴(PC1),第二主轴(PC2)和第三主轴(PC3)的奉献率分别为50.37%,30.14%和10.67%。图中H3和H4之间的距离最近,表明两者的沉积物微生物群落构成及丰度都相近。H6和H2,H6和H5沉积物微生物群落构和丰度也比较相近,但相似性小于H3和H4沉积物样品微生物组成。H1与其他5种沉积物样品微生物群落组成之间差异都比较大。这与我们基于高通量测序的微生物多样性和丰度研究中所得出的结果一致,乌海(H1)沉积物样品微生物多样性和丰度在6种样品中都是最低。
2.2 黄河沉积物样品细菌群落结构及类型
本研究6个采样点沉积物样品的36447个OTUS分属于365个门,1128个纲,2170个目,3360个科,4722个属,可见黄河内蒙古段沉积物有着比较丰富的微生物多样性。通过与Green gene database中的已知序列进行比对后,对黄河沉积物样本中的细菌群落结构有了初步的了解。黄河表层沉积物样品主要细菌种类在门纲目科属水平上的相对丰度见图3至图7。由图可见,黄河沉积物样本细菌群落在门水平上,变形菌门(Proteobacteria,相对丰度为32.39)、绿弯菌门(Chloroflexi,相对丰度为13.25%)、拟杆菌门(Bacteroidetes,相对丰度为12.16%)占据了总细菌群落丰度的50%以上。说明这三类细菌在黄河内蒙古段沉积物中为优势菌群。在变形菌门中发现的主要种类有α-变形菌纲(AlphaProteobacteria),β-变形菌纲(BetaProteobacteria),γ-变形菌纲(GammaProteobacteria),δ-变形菌纲(DeltaProteobacteria)等四种,其中β-变形菌纲占据了变形菌总丰度的36.98%,δ-变形菌纲所占相对丰度为23.86%,α-变形菌纲所占相对丰度为20.89%,γ-变形菌纲所占相对丰度为15.86%。由此可见,β-变形菌纲在黄河内蒙古段沉积物中生态位置中占据比较重要的位置。
本研究中发现的含量比较多的细菌类群还包括后壁菌门(Firmicutes,相对丰度为7.71),酸杆菌门(Acidobacteria,相对丰度为6.15),放线菌门(Actinobacteria,相对丰度为7.10),它们的相对丰度均在6%以上。本研究还发现了芽单胞菌门(Gemmatimonadetes),疣微菌门(Verrucomicrobiae),蓝细菌门(Cyanobacteria),硝化螺旋菌门(Nitrospira),螺旋体门(Spirochaetes),绿菌门(Chlorobi),梭杆菌门(Fusobacteres),浮霉菌门(Planctonycetes)等相对丰度比较少的种群。此外,在我们的比对结果中有5%的细菌16S rRNA基因序列在基因库中没找到相似序列或所占比非常少,所以将其一并划分为其他(Other),这些序列也许是至今为止尚未被发现的一些新的微生物种类或是在这个环境中从来没培养的微生物的一部分。
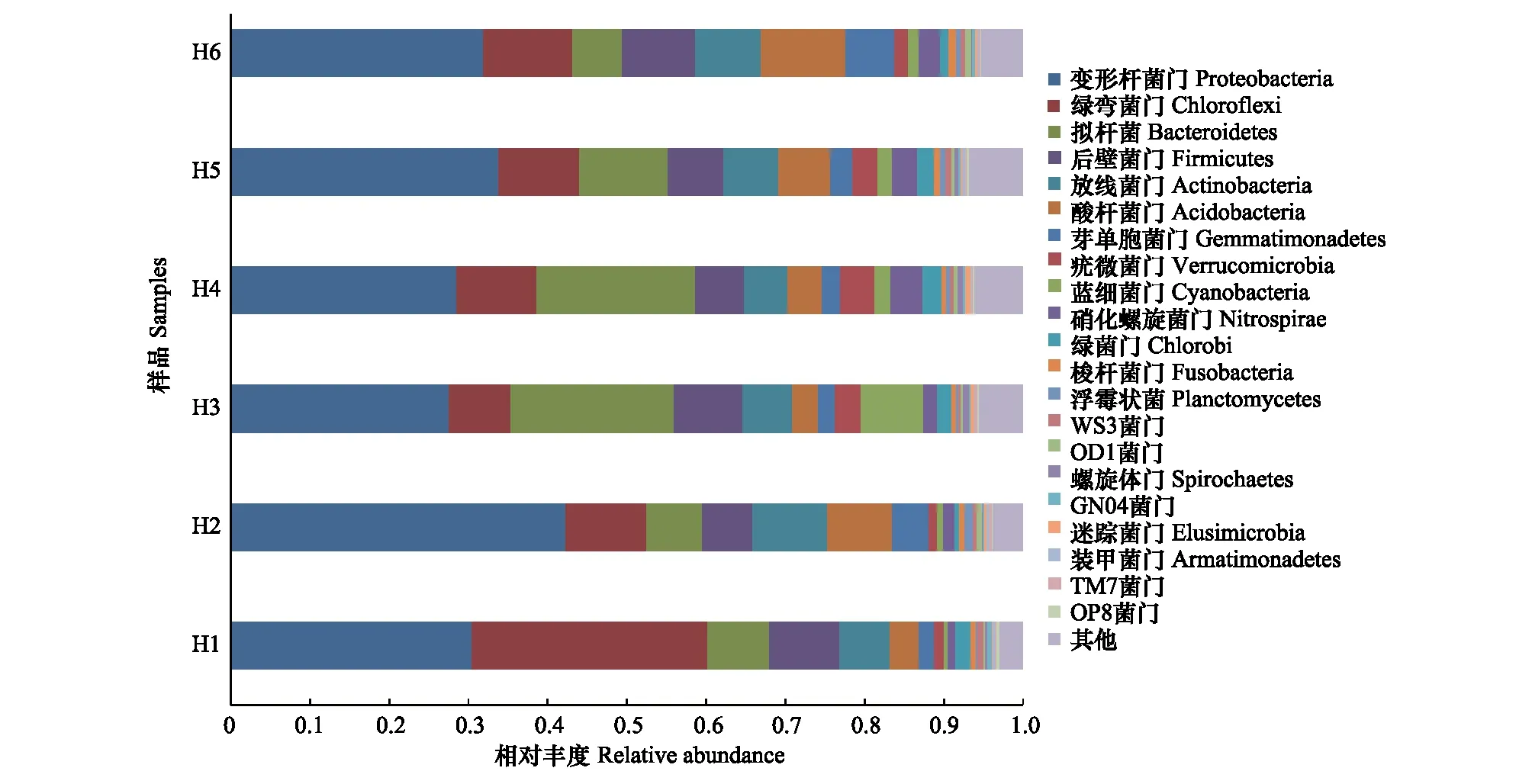
图3 沉积物样品主要细菌种类在门水平上的相对丰度Fig.3 Relative abundance of clone libraries in sediment of Yellow River
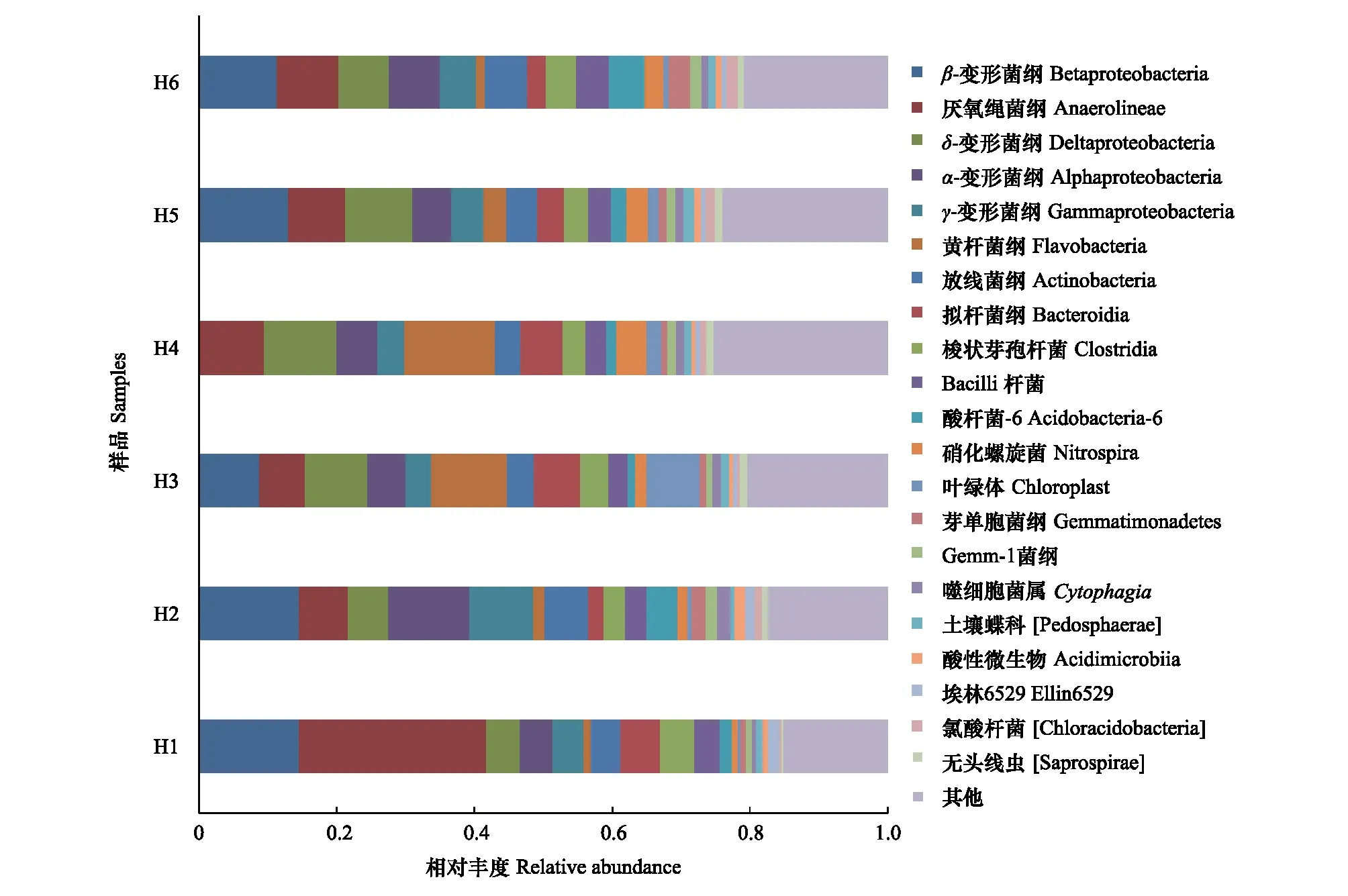
图4 沉积物样品主要细菌种类在纲水平上的相对丰度Fig.4 Relative abundance of class libraries in sediment of Yellow River
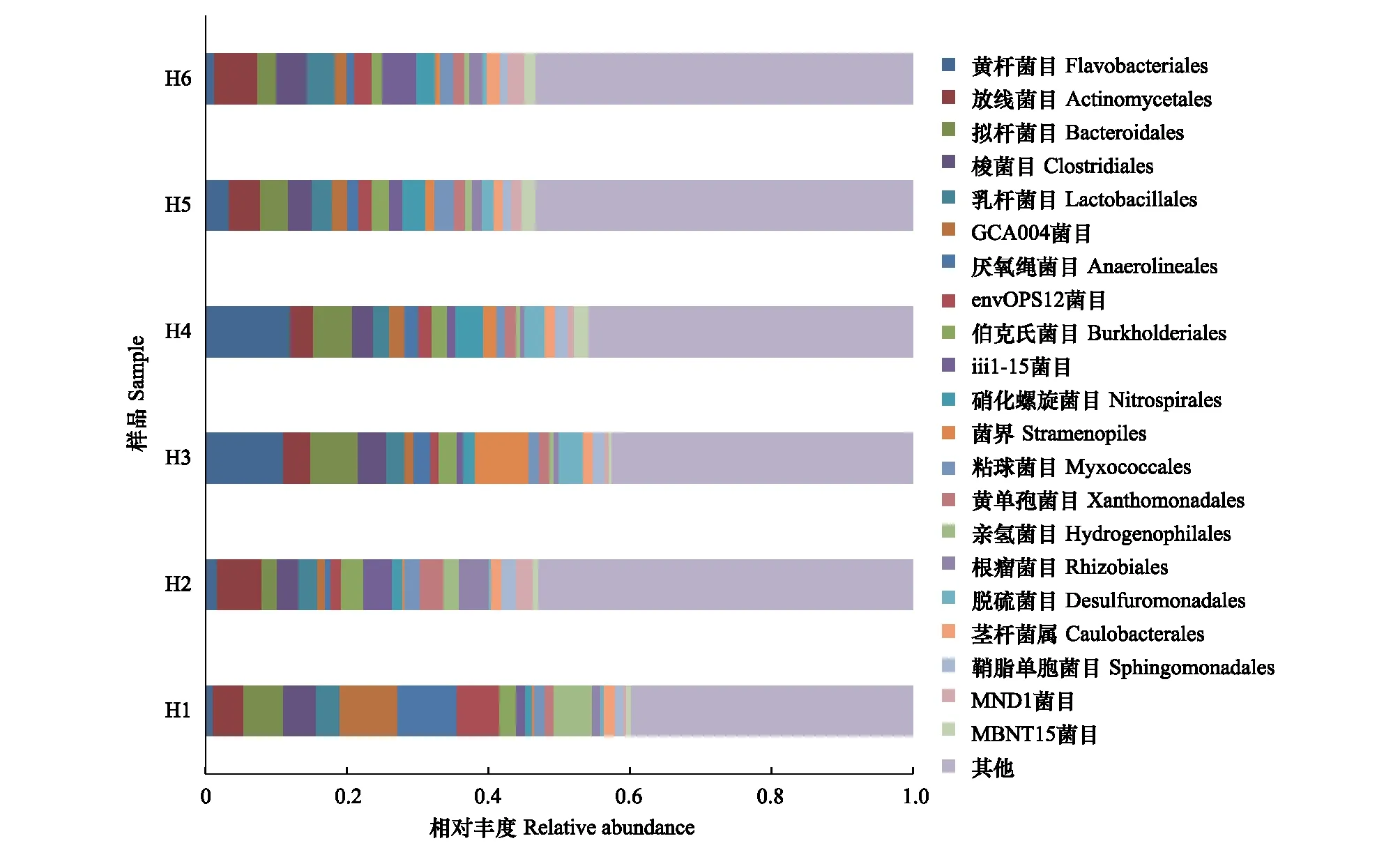
图5 沉积物样品主要细菌种类在目水平上的相对丰度Fig.5 Relative abundance of order libraries in sediment of Yellow River

图6 沉积物样品主要细菌种类在科水平上的相对丰度Fig.6 Relative abundance of section libraries in sediment of Yellow River

图7 沉积物样品主要细菌种类在属水平上的相对丰度Fig.7 Relative abundance of genus libraries in sediment of Yellow River
2.3 黄河沉积物微生物多样性与环境因子之间的关系
沉积物理化指标测定结果见表3。CEC最大值是乌海沉积物,最小值是乌拉特前旗;TOC最大值是乌海沉积物,最小值是乌拉特前旗;TP最大值是乌海沉积物,最小值是老牛湾;LOI最大值是乌海沉积物,最小值是包头。

表3 沉积物的理化参数
黄河内蒙古段表层沉积物门分类水平上的细菌群落与环境因子之间的冗余分析结果如图8所示。分析结果显示,沉积物中TOC、CEC和TP等环境因子对黄河内蒙古段沉积物细菌群落分布影响较大。有研究报道对细菌群落及其环境影响因子进行冗余分析(RDA)[3, 17-18]结果表明,发现TOC、总氮(TN)、TP等环境因子对沉积物细菌分布影响显著,与本研究所得结果基本一致。

图8 沉积物样品细菌群落与理化指标的冗余分析Fig.8 Redundancy analysis of bacterial phyla and environmental parameters in sediment sample
黄河内蒙古段表层沉积物微生物多样性指数与各形态氮和环境理化因子做相关性分析结果见表4。数据处理中使用 SPSS Statistics19.0软件对各组数据进行双变量相关性分析,得出显著性概率P值(*:P< 0.05,说明 0.05水平(双侧)显著相关)。从表4可知,黄河沉积物各样品细菌群落多样性Chao1和ACE指数与沉积物中TN、TP呈显著负相关,说明黄河沉积物细菌群落丰度的大小对氮磷等营养物质的损耗有直接关系;与沉积物中铁锰氧化态氮(IMOF-N)、碳酸盐结合态氮(CF-N)呈显著负相关,可能对可转化态氮的释放与转化有着很显著的影响。而Shannon指数仅与IMOF-N、CF-N呈显著负相关,说明微生物群落的多样性对铁锰氧化态氮和碳酸盐结合态氮的释放有促进作用。沉积物CEC的大小基本上代表了沉积物可能保持的养分,这三种指数均与CEC呈显著负相关关系,说明微生物群落的多样性会消耗沉积物养分。相关性分析结果表明,大多情况下微生物群落多样性及丰度在不同程度上会促进氮形态的转化和释放,但其过程复杂多样,因此微生物在氮形态转化中的具体作用还需进一步探索和研究。
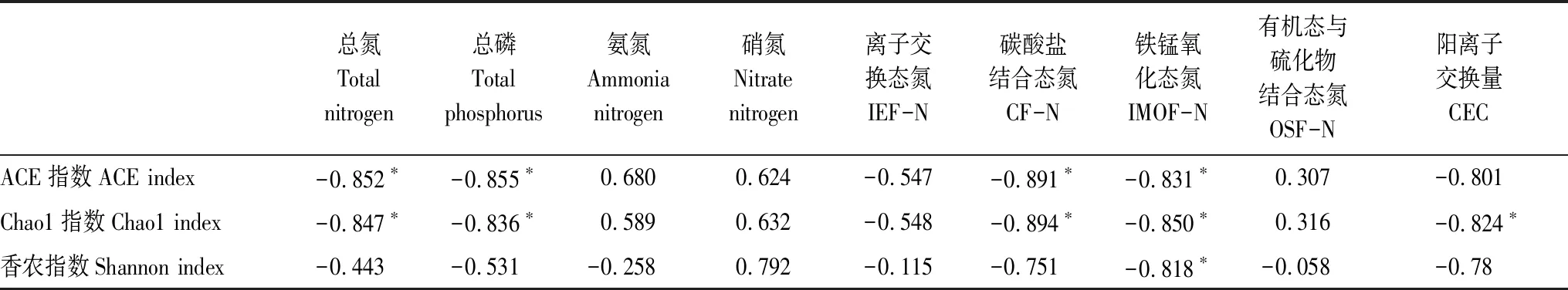
表4 多样性指数与各形态氮、环境因子之间的相关性分析
3 讨论
3.1 优势细菌组成及功能
物种多样性是维持生态系统正常功能的前提条件[19]。对6个黄河表层沉积物细菌群落组成进行研究,发现其主要由变形菌门(Proteobacteria,32.39%)、绿弯菌门(Chloroflexi,13.25%)、拟杆菌门(Bacteroidetes,12.16%),后壁菌门(Firmicutes,7.71%),酸杆菌门(Acidobacteria,6.15%),放线菌门(Actinobacteria,7.10%)组成,序列总和占全部序列的78.76%,说明这六类细菌在黄河内蒙古段沉积物中为优势菌群,表现出群落组成的丰富性。黄河沉积物细菌群落组成与之前研究的黄河三角洲湿地[20]、丹江口库区[9]、太湖[21]、北运河[22]等河流及其海洋[23]等水体的沉积物细菌组成类群相似,但又有区别。王鹏等[8]鄱阳湖典型湿地主要菌群为变形菌门、酸杆菌们、绿弯菌门、硝化螺旋菌门、厚壁菌门等; 阴星望等[9]丹江口库区表层沉积物主要菌群为变形菌门、绿弯菌门、拟杆菌门、疣微菌门、硝化螺旋菌门等;郭建丽等[24]双台子河口沉积物主要菌群是变形菌门、放线菌门、拟杆菌门、酸杆菌门以及绿弯菌门。研究表明这些沉积物细菌群落的丰富性和多样性使沉积物在营养盐循环、有机物降解、重金属形态转化等方面起着重要的生态功能[25]。
变形菌门是一大类细菌群落,在很多细菌群落研究中都具有最高的相对丰度,如南海北部和黄海西北部沉积物[26-27],但在不同海域表层沉积物基因文库中所占的比例差异较大,卡斯卡底古陆边缘次表层沉积物中变形菌门含量占文库比例高达95%[28],而在日本 Nankai 海槽 1176 站位次表层沉积物中,变形菌门仅占基因文库的 22%[29]。东海陆架表层沉积物中变形菌门占细菌文库近50%[30];崇明东滩表层沉积物变形菌门占文库的比例在 22.4%—34.6%[31]。本研究中黄河内蒙古段沉积物中主要的微生物类群是变形菌门,占文库的比例为27.51—42.28%,平均值为32.39%。
变形菌门不同纲的菌群在沉积物中的丰度变化也是微生物群落结构研究中的一个重要指标。海洋及近海沉积物中变形菌门以γ-变形菌纲为主[32];双台子河口沉积物中细菌多样性研究表明δ-变形菌纲占基因文库的 60%,为绝对优势菌群[24]。本研究区变形菌门主要包括(按丰度大小排序)β-变形菌纲(36.98%)、δ-变形菌纲(23.86%)、α-变形菌纲(20.89%)和γ-变形菌纲(15.86%)。α-变形菌纲和β-变形菌纲是淡水细菌群落中一个典型的优势类群[33-34],包括了能与植物共生的固氮细菌[35],可为土壤提供更强的固氮能力。β-变形菌纲经常利用有机物分解产生的氨气、甲烷等营养物质,β-Proteobacteria 更易存活于受污染的环境中[36],它可作为环境质量监测与评价的生态指标[27]。在黄河沉积物中β-变形菌丰度最大,推测本研究采样地点可能受到了陆源排污或其他人类活动的影响。
δ-变形菌纲包含了以其他细菌为食的细菌,对沉积物氮、磷、硫和有机质循环有重要作用[37]。细菌群落丰度指数与TN、TP、碳酸盐结合态氮(CF-N)和铁锰氧化态氮(IMOF-N)在P< 0.05水平呈显著负相关关系,说明黄河沉积物细菌群落丰度的大小与氮磷等营养物质的损耗有直接关系。
黄河内蒙古段沉积物中相对丰度第二高的类群是绿弯菌门。绿弯菌门是一类通过光合作用,以二氧化碳为碳源产生能量的细菌[38],但它们是兼性厌氧生物,因此在光合作用中不产生氧气,从而不能固氮。另有研究表明绿弯菌门在水位频繁变化的潮间带土壤含量较高,黄河沉积物中较高的绿弯菌门丰度可能与其春季水位低、夏季水位升高交替水位变化有关。黄河水体中相对丰度居于第三位的细菌类群是拟杆菌门。拟杆菌门广泛存在于各类环境中,如海洋、土壤或与人类活动的生活环境中,它是普遍存在的共栖类群。朝鲜东海沉积物中拟杆菌的丰度也较高[39],而这株菌一般来源于动物或人类粪便,因此推测本研究采样地点可能受到了陆源排污或其他人类活动的影响。这些细菌类群以纲为单位中,也主要包括变形菌纲、酸杆菌纲、黄杆菌纲、拟杆菌纲、放线菌纲等5大类群。Foulquier等[40]研究认为干湿交替的环境有利于一部分细菌的生长,永久淹没区和干湿交替区土壤的微生物群落存在结构性差异,使得永久淹没区的黄河沉积物中绿弯菌门,酸杆菌门,厚壁菌门是与淡水生态系统中不同的菌群。
黄河沉积物中还含有少量疣微菌(相对丰度2.5%),关于疣微菌门的研究还很少。疣微菌门是革兰氏阴性细菌, 在海洋动物、南极沿岸沉积物和海水等环境中存在[41], 能在厌氧条件下进行亚硝化作用。Freitas等[42]对疣微菌门在全球海洋环境的分布和多样性的研究发现,疣微菌在海洋沉积物细菌群落中占1.4%, Ⅰ型和Ⅳ型亚门是沉积环境中丰度最高的类群, 对海洋中碳的生物地球化学循环有着重要作用。
3.2 细菌群落丰度的空间格局
黄河沉积物细菌群落丰度大小为乌拉特前旗>老牛湾>临河>包头>托县>乌海。在这六个采样点中,乌拉特前旗黄河沿岸是典型的农业区,采样时间是黄河丰水期,黄河水量已经覆盖了春季种植的土地,农民使用的化肥可能会进入水体及沉积物中,在营养物质充分的条件下,导致该地区的细菌群落丰度最高。聂三安等[43]研究施肥对微生物群落结构和多样性的影响,发现施肥处理土壤中部分微生物群落的相对丰度高于不施肥处理的土壤,说明施肥能够为微生群落提供良好的生长环境。所以,农业区微生物群落丰度较高,这与本实验样品中典型农业区-乌拉特前旗的细菌群落丰度较高的结果相符。其次是老牛湾采样点,老牛湾是著名的旅游地区,多年人类活动的影响对老牛湾的水质及沉积物有一定影响,其地理位置处于峭壁围绕中,温度等自然条件相对温和,其细菌群落丰度较高。
乌海沉积物中细菌丰度和微生物多样性都是最低的一个采样点。乌海地区是产煤地区,流经乌海的黄河由于受周围煤矿及洗煤厂的影响,黄河水的颜色偏灰黑,是所采样品中生态环境较差的一个区域。本研究六个样品中乌海沉积物中CEC、TOC、TP、LOI含量最高,丰富的有机营养可能促进特定菌类在此富集,可能会限制其他很多种微生物的生长,因而使得乌海沉积物细菌多样性指数偏低,细菌群落丰度最低[31]。乌海沉积物中绿弯菌门所占含量很高(29.7%),仅次于变形菌门(30.2%);厌氧绳菌纲(Anaerolineae)在纲水平中含量最高,达到27%;在属水平中硫杆菌含量最高,达到5.5%。乌海沉积物中绿弯菌门是六个采样点中最高的采样点,这些数据说明乌海段黄河水水质与其他采样点水质存在差异,该结果与望塘污水厂尾水排口处的研究结果一致[44]。绿弯菌门是20 世纪80 年代才被认可的一个新的系统发育分支,被认为在低氧和厌氧环境中发挥重要作用,而且乌海沉积物中厌氧绳菌纲含量最高,这表明乌海沉积物是含氧量较低的沉积物环境。
在包头采样点中,δ-变形菌纲含量最高,其序列与来源与受重金属和石油烃污染的突尼斯海峡沉积物[45]的细菌序列的相似度最高,而且包头是内蒙古重要的工业城市,黄河包头段可能受重金属的影响比较大。
3.3 细菌群落空间格局的关键影响因子
研究表明,沉积物中的总磷,总氮,有机质含量和pH值等环境因子都能不同程度的影响沉积物中细菌群落组成[46]。阴星望等[9]分析丹江口库区表层沉积物细菌群落与环境因子之间的相关性发现pH值,总磷,氨氮和有机质含量为主要影响因子。本实验采用了冗余分析(RDA)对黄河表层沉积物样品细菌群落和环境因子进行分析,结果表明沉积物中的TOC、CEC和TP等环境因子对黄河内蒙古段沉积物细菌群落分布影响较大。该结果与Liu等[46],Song等[47]研究结果类似。为进一步明确环境因子和细菌群落之间的相关关系,将微生物多样性指数与环境因子之间采用SPSS Statistics19.0进行了相关分析。结果发现,微生物多样性指数与沉积物TP,TN,ECE等环境因子呈负相关关系。戴雅婷等[48]研究也发现TN,TOC是影响细菌群落组成的重要因素,这与本实验结果相符。经过分析,沉积物TP,TN,CEC含量的大小基本上代表了沉积物可能含有的养分,黄河内蒙古段沉积物细菌群落多样性指数与TP,TN,ECE等呈显著负相关关系,说明微生物群落的多样性会消耗沉积物养分。
4 结论
(1)高通量测序结果表明黄河内蒙古段6个表层沉积物样品中6447个OTUS分属于365个门,1128个纲,2170个目,3360个科,4722个属。分析覆盖度数据和稀释性曲线结果表明样品中还有很多没有分类或未培养的细菌。
(2)变形菌门是黄河表层沉积物样品中相对丰度最高的菌门,相对丰度高达32.39%。其次是绿弯菌门(Chloroflexi,13.25%)、拟杆菌门(Bacteroidetes,12.16%)、后壁菌门(Firmicutes,7.71%)、放线菌门(Actinobacteria,7.10%)、酸杆菌门(Acidobacteria,6.15%)。
(3)细菌群落丰度与环境因子之间的冗余分析结果显示,沉积物中总TOC、CEC、TP和TN等环境因子对黄河内蒙古段沉积物细菌群落分布影响较大;微生物多样性与环境理化因子相关性分析结果表明,黄河沉积物微生物多样性对氮磷等营养物质的损耗有直接关系。
