Role of alcohol in pathogenesis of hepatitis B virus infection
2020-03-13MuraliGanesanAllisonEikenberryLarisaPoluektovaKusumKharbandaNataliaOsna
Murali Ganesan, Allison Eikenberry, Larisa Y Poluektova, Kusum K Kharbanda, Natalia A Osna
Abstract Hepatitis B virus (HBV) and alcohol abuse often contribute to the development of end-stage liver disease. Alcohol abuse not only causes rapid progression of liver disease in HBV infected patients but also allows HBV to persist chronically.Importantly, the mechanism by which alcohol promotes the progression of HBVassociated liver disease are not completely understood. Potential mechanisms include a suppressed immune response, oxidative stress, endoplasmic reticulum and Golgi apparatus stresses, and increased HBV replication. Certainly, more research is necessary to gain a better understanding of these mechanisms such that treatment(s) to prevent rapid liver disease progression in alcohol-abusing HBV patients could be developed. In this review, we discuss the aforementioned factors for the higher risk of liver diseases in alcohol-induced HBV pathogenies and suggest the areas for future studies in this field.
Key words: Hepatitis B virus; Alcohol; Immunity; Oxidative stress; Liver disease
INTRODUCTION
Hepatitis B virus (HBV) infection is an important public health problem. Two billion population worldwide infected with HBV, including 257 million chronic carriers[1].The current number of chronic HBV infection cases in United States is 2.2 million[2].However, many people living with HBV are unaware that they are infected. Usually,patients with acute hepatitis B clear HBV from their blood and liver within 6 mo.However, certain factors, such as alcohol abuse, make HBV to chronically persist which put patients at a higher risk for developing fibrosis, cirrhosis, and hepatocellular carcinoma (HCC)[3-5]. The combination of HBV infection and alcohol abuse enhances liver injury progression[6,7], especially to HCC, which is 5thmost common cancer type and 2ndleading cause of cancer death in world[5,8]. The mechanisms underlying these detrimental effects of alcohol in HBV-infected patients are not fully understood and are less clear than with chronic hepatitis C virus (HCV)infection. Current treatment for chronic HBV patients is limited to antiviral medications, interferon (IFN) injections, and liver transplants. These treatments do not fully cure the HBV infection but prevent its spread to uninfected people and decrease the chance of developing end-stage liver disease. However, these medications are often largely ineffective when chronic HBV infected patients have alcohol use disorders (AUD). Elucidation of the mechanisms behind the exacerbation of HBV pathogenesis by alcohol is crucial for the development of new drugs and treatment options in alcohol-abusing HBV patients. This article reviews the current literature concerning alcohol-mediated HBV persistence by exploring ethanol-induced immune system impairment, HBV replication, oxidative stress, endoplasmic reticulum (ER) stress, Golgi apparatus fragmentation, and a higher risk of the endstage liver diseases. It also indicates the gaps in our knowledge base for future studies in this field.
INCIDENCE OF HBV INFECTION
The epidemiology of HBV infection is geographically diverse, with population prevalence, age, acquisition mode and chance of progression to chronic state being mutually interdependent[9]. In United States, about 22100 acute hepatitis B cases were reported in 2017. The prevalence of chronic HBV infection is categorized into low,intermediate and high prevalence areas based on the percent of HBV infection’s incidence: Less than 2% is observed in low-prevalence areas (United States, Canada,and Western Europe), 2% to 7% is in intermediate-prevalence areas (Mediterranean countries, Japan, Central Asia, Middle East, and parts of South America) and more than 8% is in high-prevalence areas (Western Africa and South Sudan)[9].
DEVELOPMENT OF END STAGE LIVER DISEASES IN CHRONIC HBV INFECTION
The progression to chronic hepatitis B infection enhances the risk for development end- stage liver diseases leading to increased mortality[4,5]. The hepatic steatosis induced by HBV infection is mainly caused by HBx protein by increasing the mitochondrial reactive oxygen species (ROS) levels, oxidative stress and through the interaction with liver-enriched transcription factors, hepatocyte nuclear factor 3-β,CCAAT/enhancer-binding protein α, peroxisome proliferator-activated receptor α axis, and fatty acid-binding protein 1[10,11]. Interaction between viral proteins in the liver and immune system induces hepatocyte damage, followed by tissue repair[12].This repair process causes deposition of extracellular matrix leading to progressive liver fibrosis. HBV X protein may also have fibrogenic and oncogenic effects on liver[13]. Progression to advanced fibrosis can be either rapid or slow, or sporadic based on disease stages and levels of liver inflammation and injury[14]. A recent study reported that elevated α-fetoprotein levels and hepatitis B e antigen (HbeAg)-negative hepatitis are risk factors for liver fibrosis. In addition, these authors found that interleukin (IL)-1β elevation is important for the progression of liver fibrosis during chronic HBV infection[15]. The mean age of cirrhosis onset in chronic HBV infection acquired during childhood is about 40 years, and the complications become clinically evident 3 years to 5 years later[3,16]. Cirrhosis development is 3-fold more frequent in chronic HBV patients with high viral load than in those with low viral load[17-19].HBeAg-positivity and elevated HBV DNA levels were reported as risk factors for the onset of liver cirrhosis in patients with chronic hepatitis B[20]. Liver cirrhosis is a premalignant condition that increase incidence of genetic aberrations and cellular transformations. The chronic hepatic inflammation as well as increased hepatocyte turnover found in cirrhosis lead to genetic mutations. Uncontrolled proliferation and the high rate of genetic mutations promote progression to liver cancer[21]. HBV infection is one of the major risk factors for the development of HCC. Below, we will overview the role of alcohol in progression of HBV-infection to end-stage liver disease.
ROLE OF ETHANOL METABOLISM ON VIRAL HEPATITIS
Alcohol abuse is another major health problem prevalent throughout the world. AUD is characterized by compulsive alcohol intake and a pessimistic mood when not using alcohol. The National Survey on Drug Use and Health found that 15.1 million adults and 623000 adolescents (age 12-17) had AUD. Only 6.7% of these adults and 5.2% of these adolescents received treatment. Furthermore, alcohol abuse poses an extraordinary economic burden. Excessive alcohol use cost the United States $249 billion per year[22], and alcoholic liver disease (ALD) is an escalating global problem accounting for more than 3 million deaths annually[23].
Chronic alcohol intake alters the architecture and compromises the functional capacity of the liver. Alcohol metabolism is catalyzed by alcohol dehydrogenase and cytochrome P450 2E1 (CYP2E1) to acetaldehyde and this major metabolite is the culprit for the majority of the toxic effects associated with alcohol use[24,25].Acetaldehyde is both highly toxic and carcinogenic[26]. CYP2E1 is involved in the induction of ROS, which interact with fat molecules thereby causing lipid peroxidation[27]. In addition, both acetaldehyde and CYP2E1 induce oxidative stress[28].Overall, the effect of alcohol metabolism on protein function, DNA, changes to the immune system and oxidative stress affect both hepatocytes and other liver cells.They take place under both acute and chronic exposure to alcohol and induce significant functional impairments resulting in cell death, tumorigenesis, altered cell to cell communication, and become more prone to viral infections[29,30].
Ethanol metabolism is often associated with viral hepatitis, because liver is a primary site for both hepatotropic viruses (HCV and HBV) replication and ethanol metabolism. ALD accompanied with the hepatitis virus accelerates the disease course[31]. Synergic hepatotoxic effect caused by alcohol and HCV infection increased the risk of advanced liver disease, rapid progression of fibrosis, and higher prevalence of HCC[32,33]. It has been reported that combination of HCV infection and daily alcohol intake (> 80 g) increased the risk of HCC development > 100-fold[34]. The incidence of HBV is higher among alcoholics than among the general population[35,36]. Studies has been conducted on the combined effect of alcohol and viral hepatitis in the progression of liver diseases, but the role of alcohol metabolism as risk factors in pathogenesis of HBV infection has not been studied yet[30].
CLINICAL EVIDENCE OF HBV INFECTION ASSOCIATED LIVER DISEASES IN ALCOHOLICS
Alcohol abuse pattern has wide geographical distribution depending on alcohol drinking habits in various parts of the world. As reported, about 50% of HBV carriers drink alcohol and more than 10% are heavy drinkers in Korean population[37]. A study from Taiwan reported that alcohol drinking is linked to a lower prevalence of hepatitis B surface antigen (HbsAg) alone but to higher prevalence of HBeAg among HbsAg-positive drinkers compared with nondrinkers[38]. Recently, Iida-Ueno et al[27]extensively reviewed the role of alcohol in the exacerbation of HBV infection and progression to end-stage liver diseases. Marcellin et al[39]found a strong association between alcohol consumption and mortality in HBV patients. Two prospective community-based cohort studies from Taiwan and Korea reported that alcohol consumption had an increased risk of HCC in HBsAg-positive men when compared HBsAg-positive patients with HbsAg negative patients without alcohol consumption,but relative risk was not significant[40,41]. It has been shown that chronic HBV infection potentiated by co-factors, such as alcohol consumption, may act in synergy with the virus in determining an early onset and a more rapid progression of HCC[42,43].Furthermore, the risk of HCC development is 6-fold higher in alcohol abusers[44]. In addition, according to Loomba et al[45]both obesity and alcohol have synergistic effects in increasing the incidence of HCC in HBsAg-positive men. It has been reported in cohort of Italian cirrhotic patients that the combined effect of alcohol and HBV was high risk factor for HCC (18-fold increase) than the HBV alone[34,46]. In addition, people who use alcohol for at least 15 years had enhanced the risk of liver cancer in chronic HbsAg carriers for 3-4 times[46].
Alcohol also increases the risk of fibrosis in patients with coexisting HBV[47], as well as enhances liver necroinflammatory changes in HBsAg positive patients[48]. There is an increased alteration of liver tests in HBsAg alcohol abusers[49]. In addition, selfresolved HBV infection (defined as HBsAg-negative and HBcAb-positive) can be qualified as a risk factor for developing HCC in patients with alcoholic cirrhosis[50].Interestingly, a recent study on liver disease progression in subjects with simultaneous presence of HBV/HCV dual infection and history of alcohol abuse suggests that females are at a higher risk of liver cirrhosis than males[51]. Future studies should focus on the unresolved issues, such as the influence of alcohol in inactive HBsAg carriers, immune tolerant or long-term virally suppressed patients for the risk of liver disease progression[34]. Importantly the mechanisms of synergistic effects between alcohol and HBV infection, which increases the risk of end-stage liver diseases should be the subject of extensive research[52].
HBV REPLICATION CYCLE AND ALCOHOL
Under normal circumstances, HBV behaves as a stealth virus, escaping the immune response[53,54]. HBV is an enveloped DNA virus containing a partially double-stranded relaxed circular DNA genome tropic to hepatocytes[55]. The HBV replication cycle requires binding and entry of the virus via its receptors, cytosolic transport and uncoating of the nucleocapsid, formation of covalently closed circular DNA in the nucleus, the transcription and translation of virus-specific genes, assembly of capsids and initiation of reverse transcription, followed by budding and secretion of virions and sub-viral particles as shown in Figure 1.
HBV replication cycle is a classical process, which is regulated by both host and viral factors[55-57]. Double-stranded DNA genome encodes only 7 viral proteins including DNA polymerase, capsid protein (Core), HBeAg, X protein, and three envelope proteins: LHBs (L), MHBs (M) and SHBs (S)[57,58]. A hallmark of all Hepadnaviridae is the secretion of surface proteins as sub-viral particles (for HBV,HBsAg particles) in spherical or filamentous form and HBsAg do not contain viral DNA and are non-infectious[59].
Alcoholic patients often have higher levels of HBV markers. Under experimental conditions, Larkin et al[35]found that ethanol-fed mice had up to 7-fold higher levels of HBsAg and HBV DNA compared to control diet- fed mice. HBV RNA levels were increased in alcohol-fed mice, also showing higher expression of core, surface, and X antigens in the liver. This is consistent with the higher HBV marker levels present among alcoholics and supports the idea that alcohol abuse increases HBV replication.The ability of HBV to evade and/or suppress the immune system also supports this idea, especially when the immune system is impaired by excessive alcohol consumption. Recently, based on in vitro studies, we reported that ethanol metabolism increased the HBV RNA, covalently closed circular DNA, and HBsAg in HBV transfected cells[60]. This report is in agreement with a previous study which demonstrated that ethanol significantly increased HBV replication in mice[61]. The mechanism behind the ethanol- induced HBV replication may be related to increased CYP2E1 activity and subsequent oxidative stress induction. As shown by Min et al[62]ethanol-induced overexpression of CYP2E1 significantly increased the expression of HNF-4a, a major transcription factor for the HBV core promoter, thereby increasing the HBV replication in ethanol exposed HepAD38 cells. The same authors reported that alcohol per se stimulates the HBV genome transcription by increasing the liverspecific transcription factors/nuclear receptors in an oxidative stress-independent mechanism. In addition, there are other factors, PPARa, FXRa, and PGC involved in regulation of HBV RNA transcription[63,64]. They also bind to HBV core promoter,thereby increasing the transcription of HBV pgRNA[65-67]. The above-mentioned mechanisms are attributed to ethanol-induced activation of HBV transcription[62].Recently, Lin et al[68]showed that alcoholic HBV patients have higher hepatitis B viral load. In addition, acetaldehyde affects the lipid composition of cellular membranes in lipid rafts, thereby influencing HBV infectivity[30,69]. Thus, the increased HBV replication plays a role in establishment of chronic hepatitis and/or liver end-stage disease in alcohol abusing HBV patients.

Figure 1 Schematic presentation of hepatitis B virus replication cycle. Hepatitis B virus (HBV) enters hepatocytes via hepatocyte-expressing receptors for viral entry, either sodium taurocholate co-transporting polypeptide or heparan sulfate proteoglycan. The next stage is uncoating of the nucleocapsid, which takes place in cytosol and then formation of covalently closed circular DNA occurs in nucleus. Next, the transcription and translation of HBV specific genes take place and finally, the HBV virions are released to circulation. X protein, and three envelope proteins: LHBs (L), MHBs (M) and SHBs (S). HBV: Hepatitis B virus; NTCP: sodium taurocholate cotransporting polypeptide; cccDNA: Covalently closed circular DNA; HSPG: Heparan sulfate proteoglycan; rcDNA:Relaxed circular DNA; ER: Endoplasmic reticulum; Pol: DNA polymerase; Core: Capsid protein; HBeAg: Hepatitis B e antigen.
HBV PATHOGENESIS/IMMUNOPATHOGENESIS AND ALCOHOL
The natural history of HBV has been subdivided into two types of infection. In adults,90%-95% of HBV infections is acute where immune-competent people clear the viral infection effectively[70,71]. Acute infection is characterized by inflammation and necrosis of hepatocytes and has low mortality rate (0.5%-1%)[71]. The persistence of HBsAg in blood for longer than 6 months after the initial infection is a sign of chronic hepatitis B[70]. This infection is mainly asymptomatic without any intense liver damage, but in some cases, it leads to chronic hepatitis, followed by fibrosis, cirrhosis development,and HCC. Majority of infected children aged 1-5 years, are not able to clear HBV and represent the source of chronic patients[71,72], whereas 5%-10% of HBV-infected adults are prone to develop chronic HBV infection with the mortality rate of 15%-25% from cirrhosis and HCC[71,73].
Based on the virus-host interactions, the natural course of chronic HBV infection is sub-divided into 4 stages[3,74]: (1) Immune-tolerant phase is characterized by HBeAg positivity, and high levels of serum HBV DNA due to active HBV replication[75,76].Mostly, children and young adults who are HBsAg positive for 10-30 years from the initial infection are in this phase[71]; (2) Immune clearance phase accompanied by elevated serum ALT levels and decreased HBV DNA load; and (3) Immune-control phase is characterized by low-replication, patients lose HBeAg with seroconversion to anti-HBeAg, accompanied by liver disease remission; this is typical for the inactive carrier state. However, about 20%-30% of these patients may have a viral relapse followed by reactivation phase during follow-up period[75,76].
There are very limited reports available to support the role of alcohol in HBV pathogenesis in relation to HBV markers. For example, it has been reported that alcohol abused cirrhotic patients with higher levels of serum HBV DNA are more prone to liver cancer than those with low serum HBV DNA. An increasing HBV DNA levels precipitate the progression of liver cirrhosis to HCC[77]. In another study, the synergism between HBsAg positivity and drinking were reported, suggesting a stronger influence of viral infections and alcohol drinking on the risk of liver cancer[78].In contrast, as reported by an older study, increased alcohol consumption is related to higher prevalence of HBeAg seroconversion to anti-HBe, increased prevalence of ALD and lower prevalence of chronic hepatitis[79]. Furthermore, some early studies demonstrated more frequent presence of anti-HBs and anti-HBc antibodies in blood of alcoholics than in the non-alcoholics[80-82]. In addition, it has been shown that alcohol consumption increased liver necro-inflammatory changes in HBsAg positive patients[48]and elevated liver tests[49].
ROLE OF ALCOHOL IN HBV INNATE IMMUNITY
Host cells activate innate immune response when they contact pathogen to prevent the spread of infection and to stimulate efficient adaptive immune response[71,83].Pattern-recognition receptors, through identifying the specific pathogen determinants activate innate immunity to protect against pathogens. Viral sensing, induction of type I IFNs and production of different cytokines are performed via toll-like receptors(TLRs), RNA helicases, RIG-I-like receptors, NOD-like receptors, melanoma differentiation-associated protein 5 and protein kinase R. Namely, TLR5, and TLR9 are receptors for viral DNA, TLR7 and TLR8 for single-stranded RNA, while TLR3 can bind double-stranded RNA[71,83-89].
Production of IFN type 1 -α/β and activation of natural killers (NK) cells are induced at the initial phase of viral infections. Infected plasmacytoid dendritic cells(pDC) are the main producers of IFN-α/β, while NK and natural killer T (NKT) cells produce IFN-γ. In addition to IFN-α/β, other cytokines, like IL-12 and IL-18, control viral replication[54,90-93].
HBV is relatively inefficient at inducing the anti-viral cytokines, including IFN-α/β.This appears to be due to limited sensing of HBV stealth virus combined with active suppression of innate immunity[94,95]. IFN-α/β induced interferon inducible genes(ISGs) are responsible for antiviral effects that minimize pathogenetic processes by limiting the viral production and spread[96,97]. Studies conducted on acutely infected chimpanzees as well as in humans showed decreased production of type-1 interferons and ISGs[94,97-100]. Interestingly, McClary et al[101]using transgenic mouse model or hepatoma cell lines have shown that HBV replicates in IFN-γ knockouts and IFN-α/β receptor knockouts mice at levels higher than those observed in wild type mice,implying that baseline levels of these cytokines control HBV replication in the absence of inflammation. In support to the above study, Lucifora et al[102]demonstrated that HBV elicits a strong and specific innate antiviral response (production of IFN-β and activation of ISGs) that results in a non-cytopathic clearance of HBV DNA in HepaRG cells. In contrast, in a chimeric mouse model, HBV inhibited the nuclear translocation of STAT1 in response to IFNα, thereby preventing ISG transcription in human hepatocytes[103]. The effect of ethanol on IFN-α/β innate responses and ISGs activation in HBV infection pathogenesis has not been investigated, but there are several studies which suggests that alcohol impairs IFN-α/β innate responses and anti-viral gene expressions in HCV pathogenesis[24,104-106]. Future studies should focus on understanding the effect of alcohol metabolism on IFN-α/β innate responses and ISGs activation during HBV infection pathogenesis as well as examine whether IFN-α/β therapy could be a useful strategy for HBV alcoholics.
HBV infection may elicit differential cytokine responses among various liver cell types different from hepatocytes, depending on the stage or route of infection.Guidotti et al[107]elegantly demonstrated that HBV can be controlled by immune cells in a non-cytolytic manner through the release of cytokines and other immune mediators. Both in vitro and in vivo studies showed that TNF-α, IL-12, and IL-18 are involved in controlling HBV replication in addition to IFN-γ and IFN-α/β induction[108-110]. Several cytokines control HBV transcription through liver-enriched transcription factors[111]. It has been demonstrated that IL-4, IL-6, IL-1β, and transforming growth factor-β (TGF-β) were effective in diminishing HBV replication markers[112-116]via regulating HBV transcriptional activity. However, while all these cytokines are protective during acute HBV infection, their persistence in chronic infection may cause liver inflammation.
While the effect of alcohol in modulating these cytokines in HBV infection has not been investigated, but it has been well documented that pro-inflammatory cytokines levels were increased and anti-inflammatory cytokines were decreased in ALD patients[117,118]. Taken together, it is reasonable to expect that alcohol could affect the anti-viral activity of these cytokines during acute infection and potentiate persistent liver inflammation in chronic HBV infection, thereby promoting progressive endstage liver diseases. The excessive or persistent presence of immune mediators in tissues have been recognized to play important roles in the pathogenesis of human diseases[117,118]. It is very important to conduct future studies to understand and fill the gaps in our understanding of the role of alcohol in both acute and chronic HBV infection induced cytokine responses.
Being important innate immunity component, NK cells control viruses via direct or indirect cytolytic effects, namely, through the release of cytokines, IFN-γ, TNF-α,TGF-β and IL-10[83,119]. In this regard, NK cell can directly lyse infected hepatocytes through granzyme/perforin or death receptor pathways causing the death of infected hepatocytes[119,120]. Non-cytolytic mechanisms of HBV clearance through cytokines like IFN-γ[121]also control virus in the infected liver without affecting cell integrity[119].
The antiviral capacity of NK cells in HBV-infection has been extensively reviewed by two different groups[119,120]. In this regard, NK cells efficiently inhibited HBV replication in a transgenic mice mouse model of HBV infection[122]and contributed to HBV clearance using acute HBV mouse model[123]. In chimpanzees, NK cells participated early in non-cytolytic clearance of HBV-infected hepatocytes accompanied by increase in intrahepatic content of IFN-gamma and TNF-α[107].However, subsequent experiments revealed a critical role for T cells rather than NK cells in HBV control in this model[124]. In the pre-clinical ramp-up phase of acute hepatitis B patients, it was observed an increase in the number of circulating NK cells[125,126], while activation and effector function was suppressed; this led to viral load increase[98]. There was inversive correlation between low NK cell activation and induction of the immunosuppressive cytokine IL-10, raising the possibility that HBV can actively evade immune responses[98].
NK cells display varying changes in proportion, phenotype and/or function in different studies of chronic HBV infection. The defects in NK cells are reflected in many aspects: In chronic HBV patients: (1) The proportions of hepatic and peripheral NK cells are reduced with or without changes in their subsets[127-129]; (2) There are changes in expression of activating or inhibitory receptors on NK cells[130,131]; (3) There is an increase of some molecules with negative effects, such as T cell immunoglobulin and mucin domain containing molecule-3[132]; (4) The cytolytic activity is maintained or even enhanced, which correlates with the severity of liver injury[128,129,131]; and (5)There is defect in the production of cytokines, like IFN-γ and TNF-α, making them inefficient in promotion of direct non-cytolytic antiviral roles as well as in T-cell responses[128,130-132].
Activated NK cells play a role in early HBV-infected hepatocyte clearance.However, with the progression of chronic infection, both NK and T cells can be suppressed by tolerogenic effects of hepatic ligands and cytokines which limit their antiviral efficacy[119,133]. Further studies are needed for better understanding of the factors triggering and mediating the opposing roles of NK cells in chronic HBV infection which could allow these cells to be successfully exploited as therapeutic targets[119]. Importantly, the role of alcohol on NK cell responsiveness in the HBVinfected liver has not been addressed. As reported, NK cells are impaired by alcohol[134], which ultimately affects antiviral activity of NK cells during acute HBV infection. Another possible mechanism is the alcohol-induced impairment of IFN-γ signaling which would decrease the protective ISGs gene expression. We recently reported that ethanol metabolite, acetaldehyde impaired IFN-γ signaling via the JAKSTAT1 pathway in HBV transfected cells[60]. Future studies in this topical area will improve our understanding of NK cell immunity in HBV-infected alcoholic patients.
Dendritic cells (DCs), (both conventional/myeloid DCs, mDCs and plasmacytoid DCs, pDCs) effectively connect the adaptive and innate immune responses[135].Subpopulations of DCs can be distinguished from other immune cells by specific surface markers[136,137]. pDCs play a crucial role against viral infection by producing vast amounts of type I interferon due to up-regulated TLR7 and TLR9 expression[138-140]. It has also been reported that pDCs increase the co-stimulatory and major histocompatibility complex (MHC) molecules expression on target cells enabling them to present antigen to T cells[141].
Cui et al[142]elucidated three main ways of recognition of HBV antigens by DCs.First is that HBV DNA can be recognized by DCs through TLR9, second DCs internalize HBV DNA and third, HBsAg can be internalized by DCs through the mannose receptor. DCs phagocytize HBV, process viral antigens to antigenic peptides and present them to CD4+ and CD8+ T cells[143]. DCs activate antibody-dependent cytotoxicity cells and NK cells, which stimulate these cells to secrete immunosuppressive cytokines, IL-10 and TGF-β, assisting in the induction of regulatory T cells (Tregs) with the participation of mDC to destroy HBV-infected hepatocytes[144]. To date, there have been no studies which investigated the effect of alcohol on DCs function in pathogenesis of HBV infection. There are several reports support that alcohol consumption decrease the DCs functional ability which leads to impaired adaptive immunity and gives more chances to detrimental events upon pathogens exposure[145-148]. It is possible that ethanol-induced dysfunction of DCs leads to reduction in their HBV antigen presentation property, thereby causing prolonged persistence of virus and progression to end-stage liver disease.
EFFECT OF ALCOHOL ON ADAPTIVE IMMUNITY IN HBV INFECTION
The adaptive immune response is responsible for viral clearance and disease pathogenesis during HBV infection[54,149]. Cell-to-cell interactions may play either protective or pathogenic roles, and are important for anti-viral adaptive immune response in HBV infection[150]. These immune cells are: (1) CD4+ T cells, the helper T cells, robust producers of cytokines required for the efficient development of effector cytotoxic CD8+ T cells and B cell antibody production; (2) CD8+ T cells directly recognizing virus-infected cells and responsible for HBV-infected hepatocytes clearance via cytolytic and non-cytolytic mechanisms[151,152]; and (3) B cells neutralizing free viral particles by antibodies to prevent (re) infection[153,154]and affect participation of helper T cell in HBV antigen presentation[150]. The development of antiviral immune response is typical for acute HBV infection, while chronic HBV patients do not generate efficient antiviral response[149].
ROLE OF ALCOHOL ON B CELL RESPONSES IN HBV INFECTION
Minimal information is available regarding the specificity of B cell responses to HBV,although different antibodies are routinely used to distinguish between clinical phases of infection[155]. Recently, two different groups, extensively reviewed the role of B cell responses in acute and chronic HBV infection[155,156]. It has been reported that in chronic HBV patients, B cells are capable of producing polyclonal antibodies, which targeted a range of HBV antigens, including HBcAg, HbeAg, and the large, medium and small forms of HBsAg[157]. Antibodies against HBV surface antigen and HBV core antigen are produced in acute HBV infection with different kinetics. Anti-HBs is considered as a marker of disease resolution whereas anti-HBc is marker of active or past infection[158]. Some studies showed that anti-HBc response has also been associated with acute liver damage[159,160]. Antibodies targeting HBsAg and HBeAg(anti-HBs and anti-HBe) appear later in acute infection and are associated with favorable outcomes of infection[161]. The well-identified antiviral effector function of B cells is related to their differentiation into plasma cells, which produce neutralizing antibodies, preventing entry of the virus into target cells either through steric obstruction or through direct binding to the receptor-binding site on virions[162,163].During HBV infection, only antibodies directed against the envelope protein (anti-HBs) have neutralizing activity, underscored by their ability to recognize and bind to key viral epitopes required for infectivity[164,165]. The function of B cells is not only in production of neutralizing antibodies, but they also act as potent antigen presenting cells (APCs), specifically for helper T cells[166]. During the flares of chronic hepatitis B,there is an enrichment of IL-10 producing B cells which modulate inflammatory events as well as HBV-specific T cell responses[167]. Again, there is a huge gap on the role of alcohol in terms of the regulation of B cells function and its input in HBV pathogenesis. It has been previously reported that alcohol decreased the B cell numbers and especially lowered the circulating B cell levels[168-170]. Alcohol-induced loss of peripheral B cells primarily affects certain subpopulations of cells, which develop into long-lived memory B cells critical in protection from the infection with same pathogen[171]. It is possible that alcohol may weaken the B cell immune responses by decreasing the level of B cells leading to reduction in antibodies against HBV antigens, thereby causing chronic HBV. Geissler et al[172]demonstrated that in female mice fed ethanol diet and immunized by DNA-based construct containing the pre-S2/S gene, the levels of anti-HBs were marginally reduced compared with those in control mice.
ROLE OF ALCOHOL ON CYTOTOXIC T-LYMPOCYTE RESPONSES IN HBV INFECTION
Regarding acute HBV-infection, there is less information available on B-cell response but HBV-specific CD4+ and CD8+ mediated responses become normally measurable during the period of exponential rise in HBV replication[125,126]. Capsid protein epitopes were specifically recognized by CD4+ T helper cells, whereas CD8+ T cells naturally recognize epitopes positioned within diverse HBV proteins. HBV-specific T cells are Th1 focused and vigorous in self-limiting acute infection compared to chronic infection[97,173-175]. CD4+ T cell response to HBV is vigorous, and multi-specific in patients with acute hepatitis who ultimately clear the virus, but it is comparatively weak in persistently infected patients with chronic hepatitis[176]. Many studies provided evidence for a strong relationship and association between CD4+ T cell response, acute hepatitis, and viral clearance[177-179]. As also reported, there is no effect on viral clearance and liver disease when CD4+ T cells are depleted at the peak of HBV infection in chimpanzees[124], suggesting that CD4+ T cells do not directly participate in viral clearance and tissue damage. It is possible that CD4+ T cells indirectly control HBV infection by facilitating the induction and maintenance of the virus-specific B cell and CD8+ T cell response[54].
HBV-specific CD8+ T cell response acts as the principal effector mechanism of viral clearance and liver inflammation[156]. HBV-specific CD8+ T cells are enriched within the infected liver, lyse HBV infected hepatocytes[124,180]and secrete cytokines (mainly IFN-γ) that trigger a process of non-cytolytic HBV clearance[121]and recruitment of inflammatory immune cells[122,181]. As mentioned earlier, like CD4+ T cells, CD8+ T cell response is detectable in acute HBV. But in chronically infected patients, the peripheral blood T cell response is weak and narrowly focused[182-184]. Maini et al[185]examined a relationship between the number of intrahepatic HBV specific CD8+ T cells, extent of liver disease, and levels of HBV replication in chronically infected patients, demonstrating that inhibition of virus replication could be independent of liver damage and that the functionality of HBV-specific CD8+ T cells was more important than total number of T cells to control HBV replication. Experiments in chimpanzees have shown that the viral clearance and the onset of liver disease coincide with the accumulation of virus-specific CD8+ T cells and the induction of IFN-γ, as well as ISGs in the liver[107,124].
In 2010, Chisari et al[54]clearly outlined the role of cytotoxic T lymphocyte (CTL)response in viral clearance by killing infected cells. Although CTL killing is an inefficient process, in order to kill the infected cells, a direct contact between CTLs and infected cells is required. Hence, it is not possible to kill all infected cells by CTLs because unlike HCV infection, HBV can infect as many as up to 1011hepatocytes[107,186].Therefore, although hepatitis in HBV infection is due to the cytopathic activity of the CTLs, viral clearance may require more efficient CTL functions than just killing. There are few studies which investigated the pathogenetic and non-cytopathic antiviral functions of the CTL response in HBV transgenic mice that develop an acute necroinflammatory liver disease after adoptive transfer of HBsAg specific CTL clones[121,180,181]. They found that CTLs rapidly enter the liver and recognize viral antigen which triggers two events: (1) Apoptosis of the hepatocytes that are physically engaged with CTLs; and (2) Secretion of IFN-γ which non-cytopathically inhibits HBV gene expression and replication in the rest of the hepatocytes[121,187]. It has also been reported that the cytopathic and antiviral functions of CTLs are completely independent from each other[121]. All these results suggest that CD8+-dependent cytopathic and non-cytopathic clearance of HBV are effective in the limitation of HBV viral infection[100,107,121,124]. Studies conducted in both animals and humans clearly showed that alcohol reduces the number of T- cells, changes the ratio of T- cell types,decrease the T- cells activation and function and finally, promote the T- cell apoptosis[171]. Geissler et al[172]showed in a transgenic mouse model that ethanolinduced effects on CTL activity against the middle envelope protein (MHBs) as well as on T-cell proliferation and cytokine release may partially contribute to a higher incidence of persistent HBV infection in alcoholics. Again, there are no studies directly investigating the role of alcohol in the context of CTL responses in HBV infection. It is very important to study the association between alcohol and HBV adaptive immune response for understanding of the exact mechanisms of alcoholinduced impairment of various arms of the adaptive immunity. The clarity in this matter will be useful for the development of treatment strategy for the end stage liver diseases in alcoholic HBV patients.
ROLE OF ALCOHOL IN HBV MHC CLASS I AND II ANTIGEN PRESENTATION
MHC class I antigen presentation pathway plays important role in the detection of virally infected cells by CTLs. MHC class I molecules are expressed on the cell surface of all nucleated cells and present peptide fragments derived from intracellular proteins[188]. As mentioned above, in HBV infection, CTLs expressing specific T- cell receptors are responsible for elimination of HBV-presenting hepatocytes. When the display of viral peptide/MHC class I complex on HBV-infected hepatocytes is altered,it may reduce CTL activation and thereby, suppress HBV-infected hepatocyte clearance[60]. Hence, the presentation of HBV-viral peptide-MHC class I complex on hepatocytes surface is necessary for the effective elimination of HBV infected cells.Sastry et al[189]and Khakpoor et al[190]reported that the recognition of different HBV viral peptides-MHC class I complex (HBV epitopes) are important for efficient immune therapeutic control of chronic HBV infection and that determination of these epitopes will be useful in delivering antiviral drugs or cytokines directly to virusinfected cells. As we mentioned earlier, MHC class I pathway is usually fueled by endogenous antigens whereas main source of Ag entering the MHC class II pathway is exogenous protein, which is endocytosed/phagocytosed by professional APCs[191].CD4+ T cell activation is triggered when the specific antigenic peptide is presented on MHC class II molecules. Exogenous antigens may also enter MHC class I pathway,which is called cross-presentation by DCs and macrophages. Furthermore, antigens expressed in the context of HBsAg virus-like particles can access MHC class I and class II pathways of primary DCs to elicit adaptive immune responses. In addition,Murata et al[192]reported that intrahepatic cross-presentation by DCs in the liver augments HBV-specific CD8+ T cell expansion, while concomitant or subsequent hepatocellular presentation of endogenously synthesized antigen is essential for expansion and cytolytic differentiation of HBV-specific CD8+ T cells induced by DC activation. There are two old studies which reported that strong MHC class IIrestricted CD4+ T cell response to HBV core is associated with viral clearance in acute HBV infection[177,193]. There are limited number of studies investigating the role of alcohol on MHC class I and II presentation in HBV- infected cells. Pasala et al[171]reported that immune response to the HBV vaccination yielded a smaller immune response in patients who abuse alcohol compared with healthy patients. We have recently reported that in hepatocytes, the combination of HBV and ethanol metabolites impairs proteasome function as well as IFN-γ-signaling through the Jak-STAT1 pathway and suppresses HBV peptide cleavage by immunoproteasome. It also disactivates protein loading complex components, TAP and tapasin, which are required for HBV peptide-MHC class I trafficking to the membrane, finally, affecting the expression of HBV core peptide 18-27- MHC class I complex on cell surface[60]. All these events may prevent CTLs activation to limit their ability to identify/clear HBVinfected hepatocytes resulting in liver inflammation and its progression to fibrosis and HCC. Importantly, future studies should be focused on the effects of ethanol on MHC-class II presentation, which is mainly catalyzed by effector cells, such as APCs.Overall, the combination of a weakened innate and adaptive immune response due to ethanol consumption could decrease the ability to clear HBV from the body, allowing the virus to persist chronically, followed by development of end - liver diseases, such as cirrhosis and HCC.
ALCOHOL INDUCED IMPAIRMENT OF GUT-MICROBIOME AND HBV PATHOGENESIS
The unique intestinal blood supply containing microbe-derived products and metabolites affect the composition of hepatic immune cells, immune microenvironment and the regulation of antiviral immune responses. The immune response in the liver is closely controlled by the intestinal commensal microbiota signals. The host’s ability to clear HBV is correlated with the establishment of commensal microbiota. Wu et al[194]using HBV- transfected mice, demonstrated the critical role of CD4+ T cells in HBV clearance mediated by commensal microbiota.Both human and animal studies have shown that the intestinal barrier of the gastrointestinal tract has exceptionally high permeability as a result of alcohol abuse.The tight junctions between epithelial cells in the gastrointestinal tract are also disturbed as a result of alcohol abuse, allowing bacterial substances to leak into the bloodstream[195-197]. It is possible that alcohol influenced bacterial composition due to depletion of beneficial commensal bacteria and high pathogenic bacteria colonization[198]. All these changes could contribute to impairment in the HBV clearance dependent on the establishment of commensal microbiota.
ROLE OF ETHANOL AND HBV- INDUCED OXIDATIVE STRESS IN LIVER INJURY
As mentioned earlier, ethanol metabolism in the liver can lead to an increase in the production of ROS, mainly hydrogen peroxide and superoxide anion[27]. Moderate drinking (< 50 g/d) has been shown to increase the probabilities of developing oxidative stress three-fold, while heavy drinking (> 50 g/d) has been shown to increase these odds by 13 to 24 times[199]. Few studies have investigated the role of alcohol-induced oxidative stress in HBV infection pathogenesis and associated liver injury. In addition, oxidative stress may play a key role in the progression of liver disease where alcohol consumption is associated with chronic hepatitis B[27]. Ha et al[200]reported that even moderate ethanol consumption promotes oxidative stress and liver injury in HBx transgenic mice, implying that compromised antioxidant defense increases alcohol-associated liver injury. In addition, Min et al[62]found that CYP2E1-induced oxidative stress potentiates the ethanol-related transactivation of HBV.Recently, we reported the ethanol metabolite, acetaldehyde induces lipid peroxidation, and adduction of proteins with 4-hydroxynonenal and malonaldehydes(oxidative stress markers) suppress the proteasome activity, necessary for generation of antigenic peptides for MHC class I -restricted antigen presentation. This results in decreased presentation of HBV peptide-MHC class I complex for the recognition by CTLs and limits elimination of infected cells[60]leading to persistence of HBV and subsequent end-stage liver diseases.
However, it is very difficult to dissect the role of alcohol- induced oxidative stress in HBV infection and associated liver injury since HBV has also been shown to cause oxidative stress[201-203]. In a study on 158 HBV patients and 42 healthy individuals, total oxidative stress levels were significantly higher for the patients infected with chronic HBV compared to the other groups[204]. Findings from a recent study consist of 296 chronic HBV patients suggesting that oxidative stress might be a useful indicator of the progression of HBV-induced liver disease in patients[202]. HBV has also been shown to induce oxidative stress in transgenic mice and human primary hepatocytes[205,206]. Also, HBx protein-induced ROS plays an important role in autophagosome formation and the ensuing viral replication[207]. In addition, the development of HCC in HBx transgenic mice is preceded by oxidative stress[208].Overall, both alcohol and HBV proteins induced ROS activated pathways (such as the mitogen-activated protein kinase pathway) that aid in the formation of chronic liver disease[200]. Since, alcohol and HBV act as an individual factor in causing oxidative stress, there are no studies which investigated the combined effect alcohol- and HBVinduced oxidative stress in the pathogenesis of liver injury. Hence, it is very important to conduct more studies to find out the synergistic effect of alcohol- and HBVinduced oxidative stress in the progression of end stage liver disease.
ETHANOL AND HBV INDEPENDENTLY INDUCE ER STRESS
When the protein load exceeds the protein-folding capacity of the ER, the unfolded protein response (UPR) is induced as an attempt to decrease the load. A strong,prolonged UPR leads to ER stress. Both ethanol and HBV have been shown to induce the UPR and ER stress[209-212].
It is known that ALD leads to an impaired ability of the liver to secrete serum proteins, such as serum albumin and clotting factor proteins. There is evidence to support that this impairment is caused by a malfunctioning ER, which makes sense because the ER is involved in folding and modification of proteins that will eventually be secreted. Howarth et al[213]reported that the exposure to ethanol metabolites caused ER fragmentation and increased expression of UPR genes in hepatocytes. The same group found that ethanol exposure caused ER morphological abnormalities, defects in hepatocyte secretion, and a strong UPR in zebrafish[213]. Several investigators reported that acetaldehyde and its adducts cause ER stress[214-218].
HBV infection has also been shown to cause ER stress. Li et al[219], found that the UPR was induced in HBV-infected hepatocytes. However, only specific branches of the UPR were induced, while the branch leading to apoptosis was not affected. This allowed UPR and ER stress to persist while preventing apoptosis[219]. Kim et al[220]reported that chronic HBV infection results in chronic ER stress. This chronic ER stress plays a major role in the pathogenesis of liver diseases, including viral hepatitis and liver cancer.
Since both HBV and ethanol abuse cause ER stress, these stress responses may be additive or even synergistic. It has been reported that ER stress is synergistically induced by alcohol in the presence of environmental factors, drugs, or viral infection[221]. The possible mechanism behind the alcohol- induced ER stress in HBV infection may be due to the alcohol-triggered viral replication (as we mentioned in the earlier section) by increasing HBV DNA, HbsAg, and HBx protein. These viral components have been involved in the UPR activation and ER stress, playing a role in HBV pathogenesis[212,220,222,223]. It has been reported that alcohol and anti-HIV drugs induced the ER stress and liver injury[224]. Importantly, for the effective elimination of HBV- infected cells by CTLs, the proteasome generated antigenic peptides should be loaded into MHC class I molecules and form peptide loading complex in the ER for the HBV peptide-MHC class I complex presentation on hepatocytes surface. We have recently reported that acetaldehyde suppressed the presentation of HBV peptide-MHC class I in HBV transfected cell, in part, due to the acetaldehyde-induced ER stress which leads to the impaired trafficking of this peptide complex in ER to Golgi then to the cell surface[60].
ETHANOL INDUCES GOLGI FRAGMENTATION
A stressed ER often corresponds with a stressed Golgi apparatus since proteins from the ER are trafficked to the Golgi before they are secreted from the cell. There is no well-characterized system of Golgi stress markers as exists for the UPR proteins in ER,so the best way to determine the presence of Golgi stress is to view Golgi fragmentation morphologically. Siddhanta et al[225]demonstrated that ethanol exposure leads to Golgi fragmentation and fragmented Golgi cannot function properly. A malfunctioning of Golgi along with a malfunctioning ER impairs the ability of hepatocytes to secrete serum and membrane proteins[225]. Recently Petrosyan et al[226-228]revealed that abnormalities in the Golgi apparatus function is crucial for the development of alcoholic liver injury. They reported that ethanol-induced Golgi fragmentation and disorganization of Golgi matrix proteins is one of the main contributors of Golgi scattering. They also found that alcohol-induced Golgi fragmentation alters the Golgi-to-plasma membrane trafficking. Interestingly, it has been reported that under viral infection, Golgi is partially fragmented[229]and that HCV also caused Golgi fragmentation[230]. There are no studies conducted on the role of alcohol and HBV-induced Golgi fragmentation in HBV infection pathogenesis and subsequent progression of liver injury.
CONCLUSION
The combination of HBV infection and alcohol abuse can provide detrimental consequences. To gain better understanding of the mechanisms behind this dangerous combination could save millions of lives. Many possible mechanisms explain the synergistic progression of end-stage liver disease and the development of chronic hepatitis B infection in alcohol-abused patients (Figure 2). These factors include alcohol-induced increase in HBV replication, oxidative stress and suppressed immune response, which finally leads to fibrosis and HCC development. Other potential contributors to this rapid disease progression are ER and Golgi stress, which are induced by each alcohol and HBV, but effects are synergized by the combination of both insults. While many potential mechanisms for the synergistic effects of HBV and alcohol abuse exist, most of them have not been explicitly studied and characterized.More research is required to understand the complex interactions between alcohol consumption and HBV infection. Once elucidated, these mechanisms could aid in the development of new treatments to prevent the progression of end-stage liver disease in alcohol abusing HBV patients.
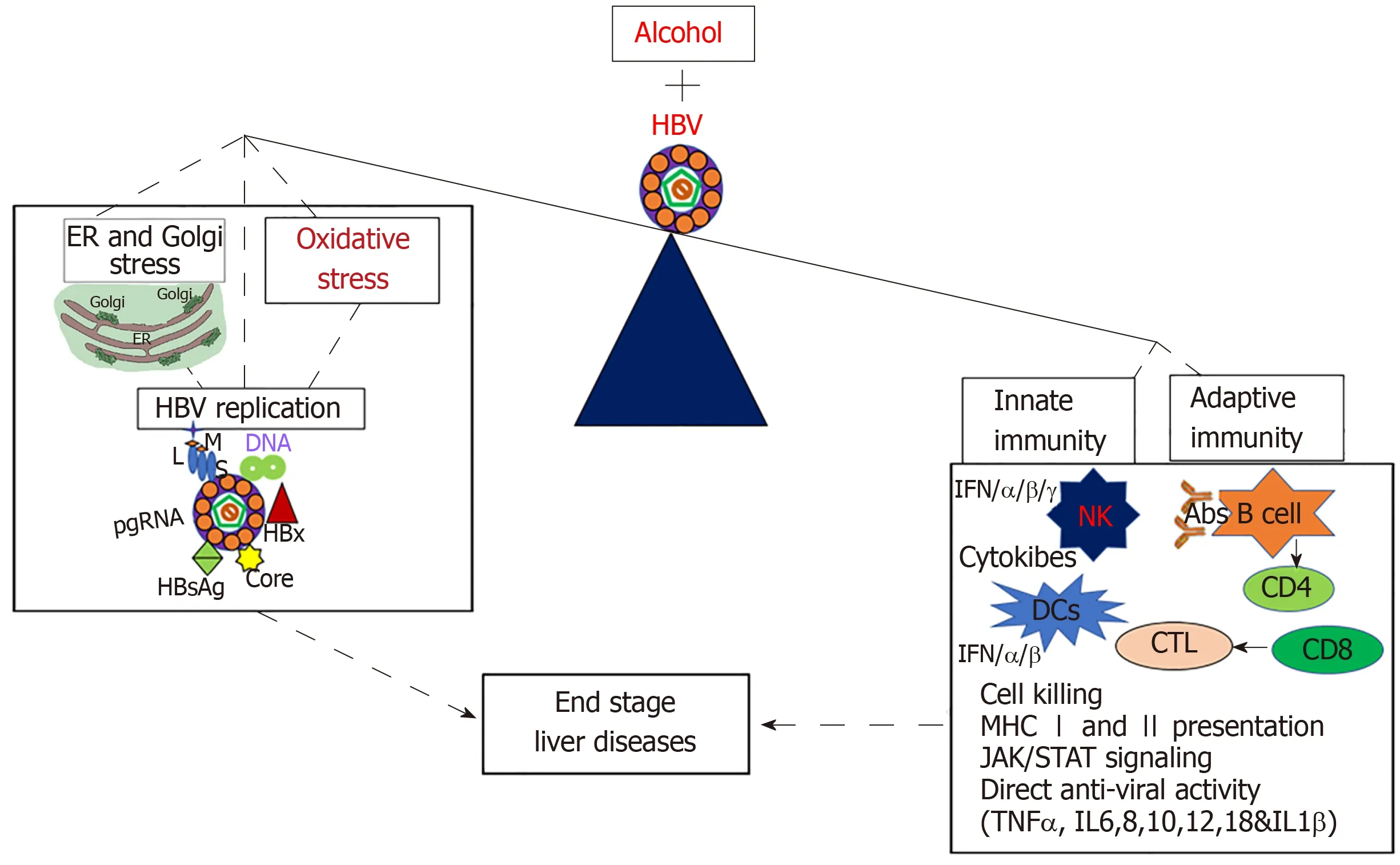
Figure 2 Mechanisms of alcohol and hepatitis B virus-infection induced liver injury. Alcohol and hepatitis B virus together increase hepatitis B virus replication,oxidative stress, and cell organelle stress (endoplasmic reticulum and Golgi stress) which ultimately suppresses both adaptive and innate immune response, thereby leading to end- stage liver diseases. HBV: Hepatitis B virus; ER: Endoplasmic reticulum; IFN: Interferon; IL: Interleukin; MHC: Major histocompatibility complex; NK:Natural killers.
杂志排行

World Journal of Gastroenterology的其它文章
- Results of meta-analysis should be treated critically
- Two case reports of novel syndrome of bizarre performance of gastrointestinal endoscopy due to toxic encephalopathy of endoscopists among 181767 endoscopies in a 13-year-university hospital review:Endoscopists, first do no harm!
- Effect and safety of mark-guided vs standard peroral endoscopic myotomy: A retrospective case control study
- Differentiation of atypical hepatic hemangioma from liver metastases: Diagnostic performance of a novel type of color contrast enhanced ultrasound
- Clinical utility of treatment method conversion during single-session endoscopic ultrasound-guided biliary drainage
- Exosomal miR-182 regulates the effect of RECK on gallbladder cancer