HPRT1 基因相关神经系统异常综合征1 例报告
2020-03-12赵培伟何学莲
赵培伟 毕 博 谭 黎 林 俊 何学莲
华中科技大学同济医学院附属武汉儿童医院(湖北武汉 430016)
次黄嘌呤-鸟嘌呤磷酸核糖转移酶(hypoxanthineguanine phosphoribosyl transferase,HPRT)是体内一种重要的酶,该酶由位于X 染色体上的HPRT 1基因编码,在体内把次黄嘌呤转化为次黄苷酸,把鸟嘌呤转化为鸟苷-磷酸[1],该基因异常可导致X 连锁隐性遗传病。HPRT 酶活性丢失的时候,次黄嘌呤及鸟嘌呤的嘌呤基团代谢为尿酸,导致高尿酸血症并出现神经系统功能异常症状[2]。HPRT 酶活性完全丧失可导致Lesch-Nyhan综合征,临床表现为高尿酸血症、舞蹈徐动症、痉挛-强直状态、精神发育迟滞、自残等症状[3]。HPRT酶活性部分缺失可导致两种表形,一种为HPRT相关的高尿酸血症(HRH),仅有高尿酸表现,不存在任何神经系统异常;另一种为高尿酸血症合并部分神经系统功能异常,但不伴有自残行为,称为HPRT相关的神经系统功能异常(HPRT-related neurological dysfunction,HRND)[4-5]。本文回顾1例利用全外显子测序技术诊断的HRND 患儿的临床资料并查询相关文献,探讨HRND的临床特点。
1 临床资料
患儿,男,3岁4个月,因独坐不稳,智力、语言发育落后来武汉儿童医院就诊。患儿6个月时竖头不稳,诊断为“发育迟缓”,接受康复治疗至今,康复进展缓慢。患儿为G1P1,出生体质量3.1 kg,Apgar评分8分,出生时未见异常。入院体格检查:营养好,神志清楚、面容未见异常;心肺腹无异常;四肢活动受限,四肢肌力4级,双手可主动抓握,但精细动作差;可短暂独坐,稳定性及平衡能力差;双下肢可部分持重,双足外翻;肌张力低下,活动及情绪紧张时四肢肌张力增高,不自主动作增多;双侧膝腱反射正常引出,Babinski征阴性。患儿父母身体健康,非近亲结婚;舅舅智力低下,生活不能自理。实验室检查:血常规未见异常,血前白蛋白56.6 mg/L,尿酸1 404.7 μmol/L,Y痕迹蛋白1.64 mg/L,乳酸脱氢酶539 U/L,乳酸脱氢酶同工酶(LD1)84 U/L,血氨基酸及尿有机酸检测未见异常。四肢肌电图、脑电图未见明显异常。头颅MRI 未见明显异常。Gesell 发育量表测定示适应性61,大运动22,精细动作45,语言58,个人社交61 分。日常生活能力评分31.5分。
经医院医学伦理审核及家长知情同意,取患儿外周血3 mL,送康旭医学检验所进行全外显子高通量测序(illumina Hiseq 2000 仪器);测序的原始数据经过组装后,采用GATK以及VarScan软件进行突变、SNPIndel的识别、注释以及统计分析,利用Annovar软件及突变数据库评定突变位点的生物学影响。结合临床表型利用HGMD、NCBI 等数据库对给出的突变数据进行再次分析,确定致病基因,并以Sanger 测序验证患者及家属的突变位点。测序原始数据经生物信息学分析注释以后得出12个可能的致病基因(SPAST,KIF1A,ADGRV1,ALDH5A1,PEX6,AHI1,VPS13B,BICD2,CBS,PEX26,DMD,HPRT1),结合患儿的家族史、临床表型(高尿酸、心肌酶谱未见升高、肌电图及脑电图无异常、无癫痫发作等)、致病基因的遗传方式,并利用SIFT、Polyphen2以及MutationTaster等软件对突变位点进行功能预测,推测HPRT1基因c.319-2A>T突变(NM_000194)可能为患儿的遗传学病因(X连锁隐性遗传),经验证患儿母亲为该位点的携带者。见图1。
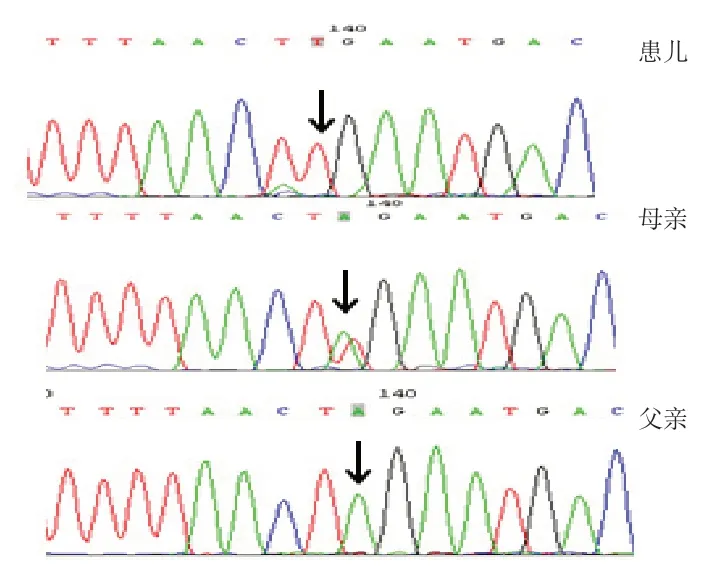
图1 患儿致病基因HPRT1 突变分析
患儿存在剪接位点突变,为进一步研究剪接位点突变对RNA加工成熟的影响,取患儿及无临床表型的正常对照外周血各0.5 mL提取RNA,利用TAKARA反转录试剂盒合成cDNA,并利用HPRT1基因外显子区域内的特异性引物进行PCR 扩增,引物序列为F:CGT CGT GATT AGT GAT GAT GAA CC,R:CCT ACAA CAA ACT TGT CTG GAA TTTC。扩增产物利用Sanger 测序分析。结果显示,与正常对照相比,患儿RNA出现两种转录本(图2A);对两种转录本进行切胶回收,测序分析发现其中一个转录本为野生型,另一个转录本缺失外显子3(图2 B、C)。提示该突变影响基因编码的RNA加工成熟,两种转录本说明突变影响HPRT酶活,但不是完全失活。患儿诊断为HPRT相关的神经系统功能异常,临床表现为高尿酸血症、神经系统功能异常(语言、智力发育落后,肌力、肌张力异常),但无自残行为。
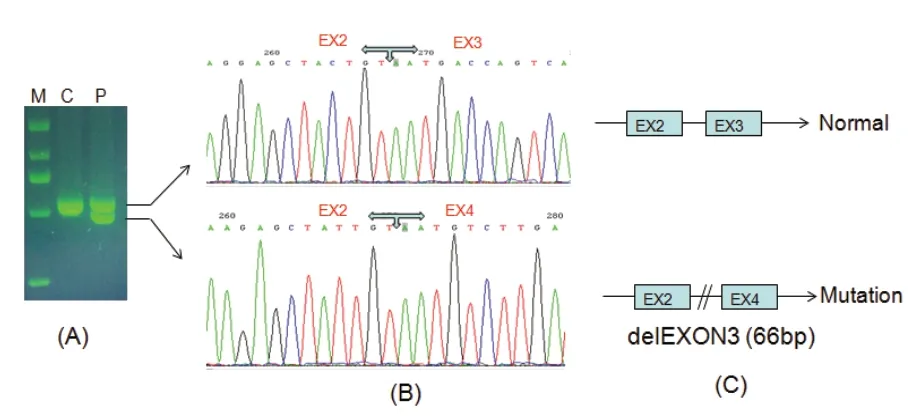
图2 HPRT1 基因c.319-2A>T 剪接位点突变的功能分析
2 讨论
HPRT 1基因位于X 染色体q 26.3 区域,全长40.5 kb,包含9 个外显子,编码含有218 个氨基次黄嘌呤-鸟嘌呤磷酸核糖转移酶(HPRT),该蛋白把次黄嘌呤转化为次黄苷酸,把鸟嘌呤转化为鸟苷-磷酸[6]。HPRT1基因缺陷影响酶的活性,次黄嘌呤在体内更多转变为尿酸。而人体内嘌呤核苷酸的合成有两种途径:从头合成途径以及补救合成途径[7]。脑组织中仅存在补救合成途径,而HPRT是补救途径中的关键酶,因此HPRT 1基因异常既能导致高尿酸血症,也可能影响神经系统的功能[8]。HPRT 完全失活导致Lesch-Nyhan综合征,临床表现为高尿酸血症、舞蹈徐动症、精神发育迟滞等神经系统功能异常并伴有自残等症状。HPRT酶活性部分缺失可导致HPRT相关的HRH;或导致HRND,临床表型为高尿酸血症合并部分神经系统功能异常,但不伴有自残行为。Lesch-Nyhan综合征以及HRND患者在中国人群中罕见报道,发病率不详,其在欧洲发病率约为1/40 万~1/20 万[8-9],为X连锁隐性遗传,临床常被误诊为脑瘫。
本例患儿6个月时竖头不稳,四肢活动受限,四肢肌力4级,双手可主动抓握,精细动作差;可短暂独坐但不稳,双足外翻;活动及情绪紧张时,四肢肌张力增高,不自主动作增多;肾功能检查提示尿酸显著升高;四肢肌电图、脑电图未见明显异常,头颅MRI 未见明显异常;发育评估落后;全外显子测序及突变筛选发现HPRT1基因存在c.319-2A>T剪接位点突变。结合表型及遗传学检测,患儿疑似Lesch-Nyhan综合征。再次随访时发现,患儿无自残行为,最终确诊为HRND。RNA水平也证实该突变影响RNA加工,患儿体内存在两种剪接异构体,分别为野生型和缺失外显子3的突变体。存在两种异构体,提示:①突变影响了正常HPRT 酶的表达和活性,导致高尿酸血症及神经系统功能异常;②存在野生型说明HPRT酶活性没有完全丧失,还存在部分活性,为患儿诊断为HRND 提供了功能学证据。
目前文献及数据库报道的HPRT 1基因突变位点超过600 个。复习相关文献发现,外显子3 可能为该基因的突变热点[5,10],且外显子3 编码的氨基酸属于HPRT蛋白的CpG岛区域,而一般CpG岛区域是一些基因的重要调控区域[11],因此该区域的多数突变可导致酶活性完全丧失,临床表现为Lesch-Nyhan综合征。曾有研究纳入83例HPTR缺陷亚洲患者,探究基因型和表型的关系发现,单个碱基的错义突变可导致Lesch-Nyhan综合征、HRH以及HRND,而无义突变则全部导致Lesch-Nyhan综合征;插入或缺失的移码突变导致Lesch-Nyhan 综合征(仅1 例除外);剪接位点突变绝大多数导致Lesch-Nyhan综合征,少数可导致HRND[12]。本例患儿为剪接位点突变,但目前诊断为HRND。由于该类疾病是一种连续的疾病谱,需密切关注患儿病情变化。
综上,由于HRND或者Lesch-Nyhan综合征在人群中发病率较低,临床表现为高尿酸血症及不同程度的神经系统功能异常。因此,临床上应对高尿酸血症特别是合并神经系统异常者进行HRND 致病基因或NGS分析,以协助明确诊断。
