Pd@C2N单原子催化剂用于HCOOH分解制氢的第一性原理研究
2020-03-11秉琦明刘靖尧
秉琦明,刘靖尧
(吉林大学理论化学研究所理论与计算化学实验室,长春 130023)
1 引言
氢能是一种可再生且无污染的重要新型能源[1].甲酸(HCOOH)因其能量密度高、产氢条件温和、无毒性、可再生等优点,被认为是一种有前途的储氢化合物[2-4].目前,用于HCOOH分解制氢的催化剂多数以贵金属(如Pd 和Pt)作为主要活性组分[5-10],然而这些贵金属催化剂较高的成本和不够理想的反应选择性阻碍了它们的广泛应用.许多研究证明,将催化活性金属组分缩小为孤立的单原子是提高催化性能同时降低成本的一种有效途径[11-15].另外,二维材料由于表面吸附位点丰富、比表面积大、物理化学性质独特等,常被用作单原子催化剂的载体.因此,将贵金属单原子与二维材料结合来设计用于HCOOH分解制氢的高效单原子催化剂值得进一步探索与研究.
最近,Baek等人成功制备了一种新型二维材料C2N,其单层所具有的周期性分布空穴和不饱和N原子为过渡金属提供了理想的结合位点[16-18].Jiang等人的研究表明,多种过渡金属单/双原子可与C2N空穴中N原子紧密结合,形成的催化剂具有优良的电催化氧还原活性[19].在我们以前的工作中[20],研究了C2N负载的Nix(x=1 -3)单原子/团簇表面上HCOOH分解制氢的反应机理,发现Nix@C2N催化剂具有优于传统的Pd基催化剂的催化性能.本研究以单层C2N为载体,将单原子Pd吸附于C2N的空穴中形成Pd@C2N单原子催化剂,并采用第一性原理密度泛函理论(DFT)计算,研究了HCOOH在Pd@C2N表面上的吸附和分解反应机理,并与传统贵金属催化剂(如Pd(111))以及Nix@C2N的反应活性与选择性进行了比较.
2 计算参数
计算使用基于密度泛函理论的VASP软件包完成[21,22],计算中使用缀加平面波(PAW)方法[23,24],交换相关能使用GGA-PBE方法[25],单电子波函数截断能为400 eV,所有计算都包含了自旋极化.如图1a所示,基于优化的C2N元胞结构构建了2 ×2 超胞模型,真空层设为15 Å以确保周期性模型之间的相互作用足够小.在结构优化和过渡态搜索中,体系所有原子完全驰豫,布里渊区积分采用Monkhorst-Pack 方法,k 点网格密度为5 ×5 ×1,过渡态使用含有8 个插入点的CI-NEB方法来确定[26,27].所有得到的中间体和过渡态结构均通过频率分析来验证,局域极小点对应全部实频而鞍点对应一个虚频.Pd原子吸附能(Eads)定义为:
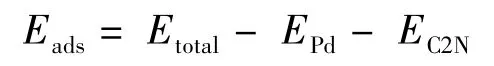
其中,EPd、EC2N和Etotal分别表示Pd原子、C2N表面和Pd@C2N体系的总能量.为了进一步验证单原子催化剂Pd@C2N的稳定性,我们开展了NVT系综下的从头算分子动力学(AIMD)模拟,时间步长为1 fs.
3 结果与讨论
3.1 Pd@C2N的结构与性质
优化后的C2N元胞含有12 个C和6 个N原子,晶格参数为a=b=8.32 Å,能带隙为1.66 eV,与报道的实验值8.30 Å和1.67 eV符合良好[16].如图1b所示,Pd原子在C2N表面的空穴内与两个N原子结合,Eads为-3.14 eV,Pd -N键长为2.12 Å.根据Bader电荷分析结果,吸附后的Pd原子带有+0.58 |e|的正电荷,从差分电荷密度图(图1c)可以明显地看出吸附后Pd 原子的电荷向C2N表面空穴中的N原子转移.
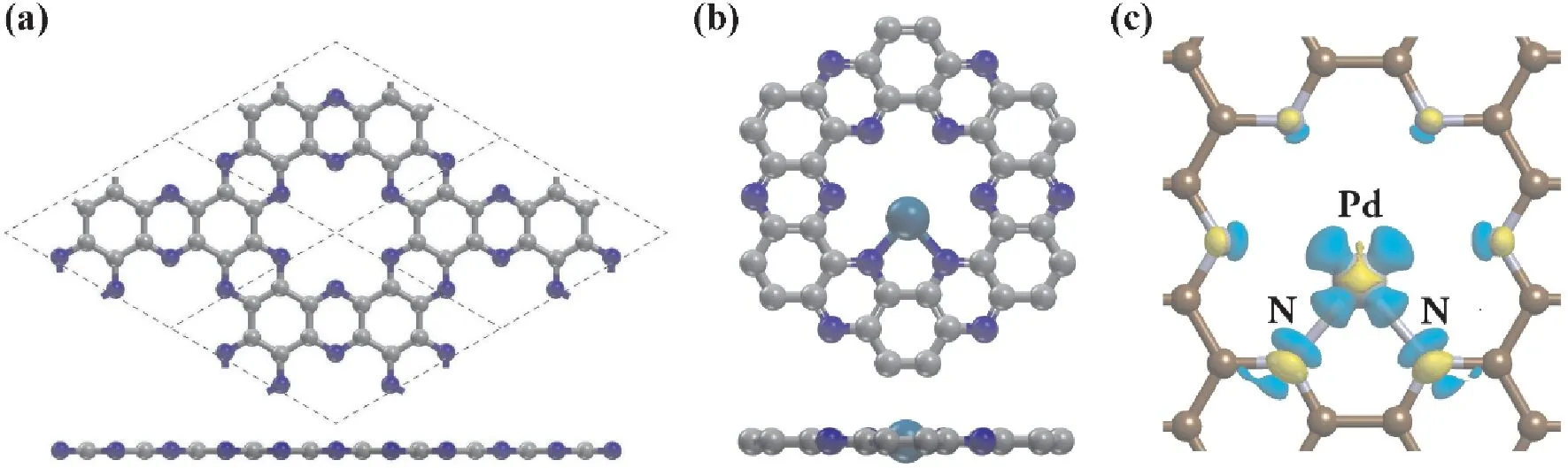
图1 (a)C2N(2 ×2)超胞模型.灰色为C原子,蓝色为N原子.(b)Pd@C2N单原子催化剂的优化结构.青色为Pd 原子,灰色为C原子,蓝色为N原子.(c)Pd@C2N的差分电荷密度图,黄色等高面代表获得电子,蓝色等高面代表失去电子.Fig.1 (a)Optimized 2 ×2 C2N supercell model.C atoms are gray and N atoms are blue.(b)Optimized structure of Pd@C2N single-atom catalyst.Pd atom is cyan,Catoms are gray and N atoms are blue.(c)charge density difference plot of Pd@C2N.In the plot,the yellow isosurfaces correspond to charge gain while the blue ones correspond to charge lost.
AIMD模拟验证了Pd@C2N具有良好的热稳定性.如图2 所示,在NVT系综和1000 K的条件下,Pd@C2N的体系总能在5 ps内保持稳定,C2N载体在模拟过程中也始终保持平面构象,且Pd原子始终结合于C2N空位中而不发生聚集,说明单层C2N能够牢固地吸附Pd 原子,是一种理想的单原子催化剂载体.
3.2 HCOOH在Pd@C2N上的吸附
如图3 所示,HCOOH可在Pd@C2N的Pd原子顶位发生吸附,吸附能为-0.22 eV,Pd -O键距离为2.35 Å.值得注意的是,HCOOH中的羟基H原子在吸附后指向了与Pd 原子相对的N原子,N-H距离为1.67 Å.通过Bader电荷分析可以发现,该N原子的电荷为-1.13 |e|,而羟基H原子电荷为+0.64 |e|,可见C2N的N原子与HCOOH中的羟基H原子之间存在着明显的静电吸引作用.
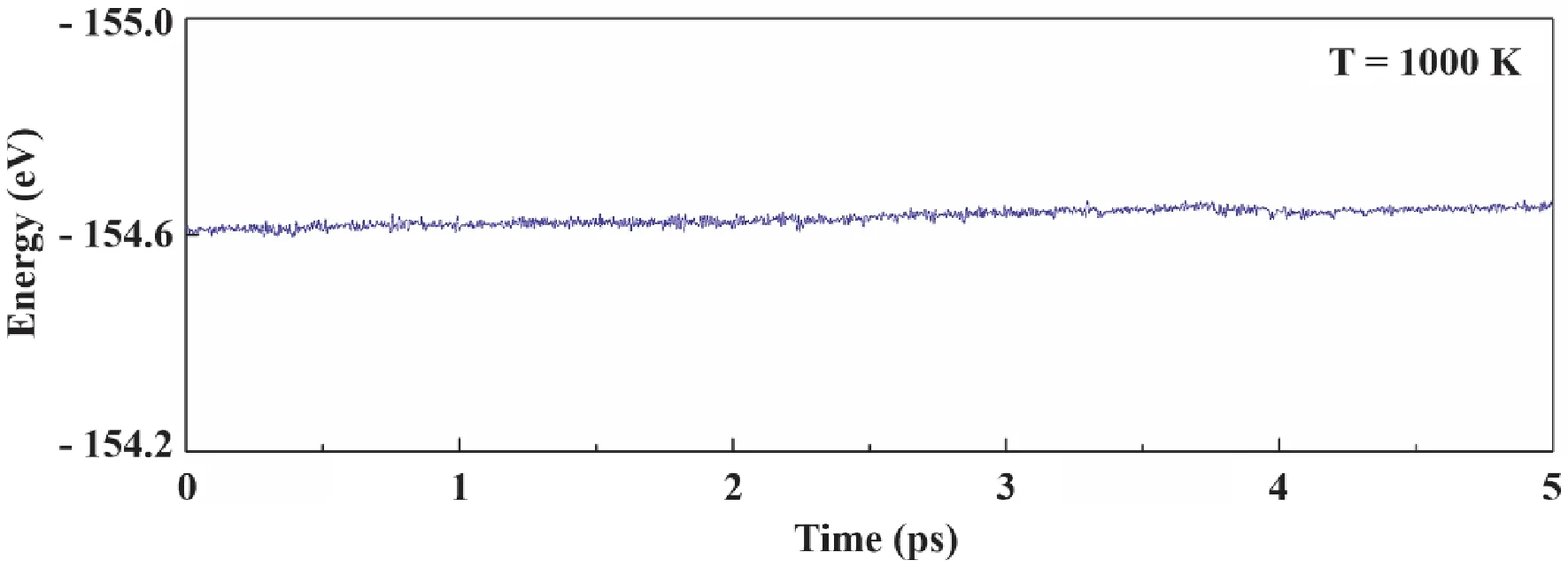
图2 单原子催化剂Pd@C2N热稳定性的从头算分子动力学模拟结果.Fig.2 AIMD simulation on the thermo-stability of Pd@C2N single-atom catalyst.
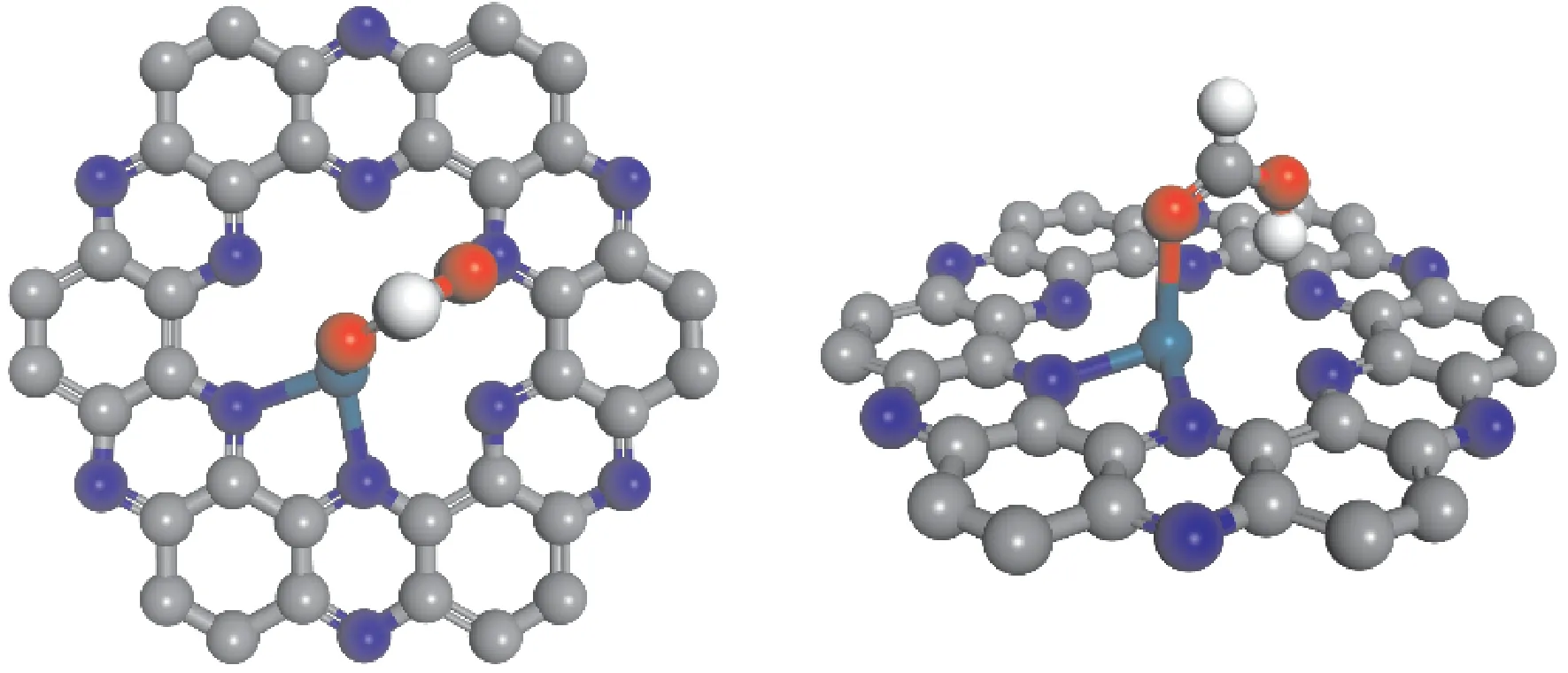
图3 HCOOH在Pd@C2N表面吸附的俯视图与斜视图.青色为Pd原子,灰色为C原子,蓝色为N原子,红色为O原子,白色为H原子.Fig.3 Top and oblique views of HCOOH adsorbed on Pd@C2N surface.Pd atom is cyan,C atoms are gray,N atoms are blue,O atoms are red and H atoms are white.
3.3 HCOOH在Pd@C2N上的分解反应机理
如图4 所示,HCOOH在吸附于Pd@C2N表面后,可通过甲酸盐路径(HCOO Path)或羧基路径(COOH Path)生成H2和CO2目标产物,反应路径中对应的中间体和过渡态结构分别在图5 和图6 中给出,Pd@C2N表面和气态HCOOH分子的总能量选作反应路径的零点.
在甲酸盐反应路径中,吸附的HCOOH分子首先在Pd和N原子的协同作用下发生解离(TS1,图5a),其中,羟基H原子吸附到C2N表面的空穴N原子上,HCOO物种以双齿构型吸附到Pd原子上(IM2,图5b).接着,HCOO物种在Pd 原子顶位发生翻转(TS2,图5c),生成单齿吸附构型的HCOO'(IM3,图5d).然后,HCOO'经过解离(TS3,图5e)生成CO2和吸附于Pd原子上的H物种(IM4,图5f).最终,分别吸附在N和Pd原子上的H物种结合生成H2.
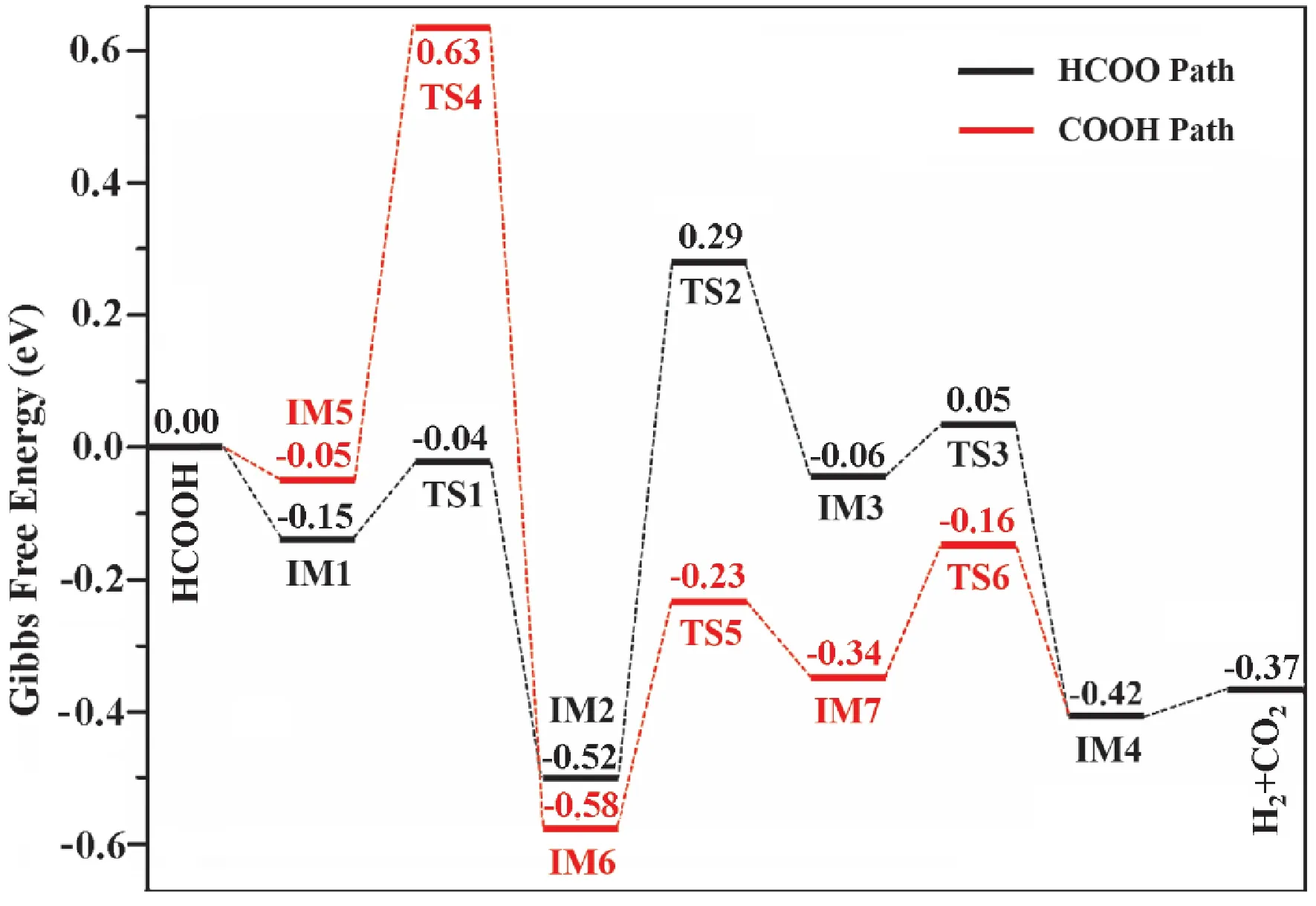
图4 HCOOH在Pd@C2N表面脱氢生成H2和CO2的反应路径.黑色:甲酸盐路径(HCOO Path),红色:羧酸路径(COOH Path).Fig.4 Reaction paths of HCOOH dehydrogenation on the Pd@C2N surface.Black line:formate path(HCOO Path),red line:carboxyl path(COOH Path).

图5 甲酸盐路径反应中间体和过渡态结构.青色为Pd原子,灰色为C原子,蓝色为N原子,红色为O原子,白色为H原子.Fig.5 Structures of intermediates and transition states in the formate path.Pd atom is cyan,C atoms are gray,N atoms are blue,O atoms are red and H atoms are white.
HCOOH的羧基反应路径从次稳态HCOOH吸附结构进行,此时吸附的HCOOH分子的α-H指向N原子,而羟基H则背离C2N表面(IM5,图6a).从该初始吸附结构出发,HCOOH首先发生裂解(TS4,图6b)生成COOH和H物种,两者分别吸附于Pd和N原子上(IM6,图6c).接着,COOH物种的羟基发生翻转(TS5,图6d),得到羟基H指向Pd的COOH'物种(IM7,图6e).然后,COOH'经过分解(TS6,图6f)生成CO2和吸附于Pd 原子上的H物种.最终,分别吸附在N和Pd原子上的H物种结合生成H2.
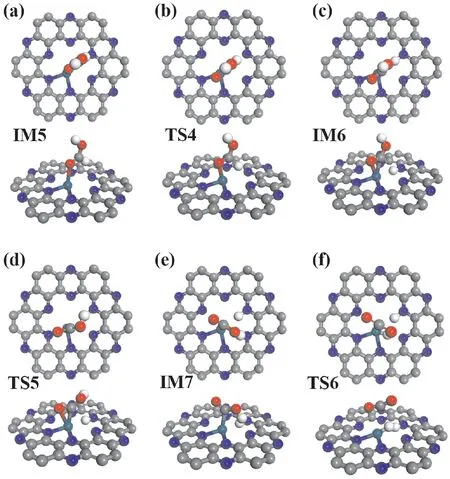
图6 羧酸路径反应中间体和过渡态结构.青色为Pd原子,灰色为C原子,蓝色为N原子,红色为O原子,白色为H原子.Fig.6 Structures of intermediates and transition states in the carboxyl path.Pd atom is cyan,Catoms are gray,N atoms are blue,O atoms are red and H atoms are white.
除了通过甲酸盐和羧酸路径生成H2和CO2,HCOOH还可能通过羧酸脱水路径(HCOOH →COOH+H →CO+H2O)或醛基脱水路径(HCOOH→HCO+OH→CO+H2O)生成H2O和CO副产物.然而,如图7 所示,Pd@C2N表面的羧酸脱水路径中COOH解离能垒较高(1.83 eV),同时醛基脱水路径中的HCO和OH中间体难以在该表面稳定共吸附,因此这两种副反应路径都难以发生,说明Pd@C2N对HCOOH分解制氢反应具有良好的选择性.
我们可以通过能量跨度模型(energy span model)[29]来衡量这两种路径的反应活性:


图7 (a)HCOOH在Pd@C2N表面上的羧酸脱水反应路径.(b,c)反应过渡态(TS7)和中间体(IM7)结构.青色为Pd 原子,灰色为C原子,蓝色为N原子,红色为O原子,白色为H原子.Fig.7 (a)Carboxyl dehydration path of HCOOH on the Pd@C2N surface.(b,c)Structures of transition state(TS7 )and intermediates(IM7).Pd atom is cyan,Catoms are gray,N atoms are blue,O atoms are red and H atoms are white.
其中,δGspan是能量跨度,TDTS(TOF-determining transition state)是决定速率过渡态,TDI(TOFdetermining intermediate)是决定速率中间体,ΔGr是反应的自由能变化.根据该能量跨度模型求得甲酸盐(HCOO)反应路径的δGspan为0.81 eV,而羧酸(COOH)反应路径的δGspan为0.84 eV,说明在Pd@C2N表面上HCOOH更容易通过甲酸盐路径生成氢气.另外,根据已有文献报道,Ni1@C2N、Ni2@C2N和Ni3@C2N表面生成氢气的δGspan分别为0.87,0.79 和1.23 eV[20],Pd(111)表面生成氢气的最低δGspan为1.33 eV[28],通过比较这些催化剂的δGspan值可以得到如下催化活性顺序:Ni2@C2N >Pd@C2N >Ni1@C2N >Ni3@C2N>Pd(111).上述研究结果表明,Pd@C2N具有比Pd(111)更好的催化活性与选择性.
4 结论
采用DFT方法设计了Pd@C2N单原子催化剂,研究了HCOOH在Pd@C2N表面上的吸附和分解制氢反应机理.结果表明,HCOOH主要通过甲酸盐路径生成氢气,通过最优反应路径的能量跨度可以判断Pd@C2N具有比Pd(111)表面更好的催化反应活性.同时,Pd@C2N对于HCOOH分解制氢反应具有良好的选择性,不易产生可能导致催化剂中毒的CO和H2O的副产物.因此,单原子催化剂Pd@C2N具有作为甲酸脱氢催化材料的应用潜力.本研究工作可为新型高效甲酸分解制氢催化剂的开发提供基础的理论指导和设计思路.
