基团贡献状态方程的开发与热力学模型参数的理论预测
2020-03-10屈绍广王昶昊施云海彭昌军刘洪来胡英
屈绍广,王昶昊,施云海,彭昌军,刘洪来,胡英
(1 华东理工大学化学与分子工程学院,上海200237; 2 华东理工大学化工学院,上海200237)
引 言
热力学模型(包括活度系数模型[1]和状态方程[2])除为过程模拟提供可靠的相平衡和热力学性质外,还可与其他理论结合构筑黏度模型[3]和界面张力模型[4],并为关联核磁共振化学位移提供广泛的可能性[5]。当今化工设计软件Aspen、ProII 和ChemCAD等都嵌入了不同的活度系数模型和状态方程,成为过程模拟、工艺开发、控制优化等的强大工具。20世纪后期,不同热力学模型相继问世,如统计缔合流体理论(SAFT)[6]、微扰链统计缔合流体理论(PCSAFT)[7]、软势统计缔合流体理论(soft-SAFT)[8]、极性统计缔合流体理论(polar-SAFT)[9]、截断微扰链统计缔合流体理论(tPC-PSAFT)[10]、变阱宽微扰链统计缔合流体理论(SAFT-VR)[11]等,模型中均具有反映分子结构特征的模型参数。与传统的立方型状态方程利用临界参数或偏心因子确定参数的方法不同,这些模型中的参数需利用流体的pVT数据拟合得到。当模型应用于混合物时,还需要混合物的相平衡数据以确定能体现不同分子间相互作用能的可调参数。
要赋予分子热力学模型具备预测多元系热力学性质的能力,使模型真正成为海量pVT和相平衡数据的“存储器”,关键是如何获得用于描述物质和系统特征的模型参数。目前的策略一是建立基团贡献(GC)状态方程(EOS),二是探索热力学模型参数的理论预测方法。本文在概括基团贡献状态方程与热力学模型参数理论预测进展的基础上,重点以开发的变阱宽方阱链流体状态方程(SWCF-VR)[12]为例,简述基团贡献状态方程构建的一般方法与热力学模型参数理论预测的基本思路,并以若干实例展示方法与思路的具体效果。
1 基团贡献状态方程
赋予分子热力学模型预测功能的首选方法是采用基团贡献法,即利用基团参数计算分子热力学模型中的参数。目前构筑的热力学模型均具有一定理论背景,原则上都可实现基团贡献化,如GCSAFT[13]、GC-PC-SAFT[14]、GC-PPC-SAFT[15]、GCmPC-SAFT[16]等。这些GC-EOS 已被拓展到不同系统,其基团贡献的数据在不断丰富中。
在构筑SAFT 型EOS 中的硬球成链项时,多数模型只考虑相邻链节间的相关性。本文则进一步考虑了相间链节的相关性[17],并结合计算机模拟结果建立了一个新的硬球链流体模型,它既适用于由相同性质的链节组成的均核硬球链流体,也适用于由不同性质的链节组成的非均核硬球链流体混合物,且不限链节的成链方式。随后,以方阱(SW)色散贡献为微扰,构筑了能描述实际系统热力学性质的方阱链流体状态方程(SWCF-EOS)[18-20],并结合二阶微扰理论和积分方程方法提出了一个变阱宽方阱链流体状态方程SWCF-VR[12,21-23]。
假定系统由N个分子组成,每个分子由r个直径为σ的硬球链节组成,链节间具有方阱作用,其阱深为ε,对比阱宽为λ,则系统的压缩因子z(SWCFVR)可表示成
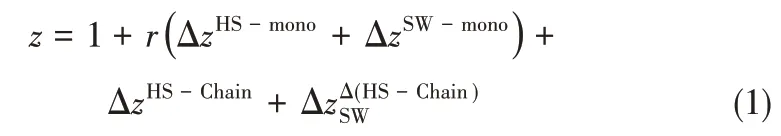
式中,上角标HS-mono 表示硬球单体对压缩因子的贡献,SW-mono 表示方阱单体的贡献,HSChain 表示硬球成链的贡献,Δ(HS-Chain)表示方阱作用对成链的影响。SWCF-VR 共有四个模型参数:链节数r、链节直径σ、方阱阱深ε和对比阱宽λ。
与所有其他模型一样,SWCF-VR 在应用时需要纯物质pVT数据确定模型参数。为赋予SWCFVR 预测功能,借鉴文献[13,24]的研究方法并假定:分子由若干不同基团组成,分子的性质则是不同基团性质的贡献,相同基团在不同物质中的贡献相同。此时,SWCF-VR 中的模型参数则与组成分子的基团参数有关,基团i对分子参数r、σ、ε和λ的贡献分别是ri、σi、εi和λi,则

基团参数可采用逐一优化的方式获得,首先利用液态直链烷烃C3~C9的密度数据得到基团—CH2—和—CH3的贡献值。在此基础上,利用C3~C9的液态烯烃的密度数据获得CH2====CH—基团的贡献值。类似地,可得到—OH,—C====O—,—Cl,—Br 和—I 等不同基团的贡献值,部分基团参数见表1。
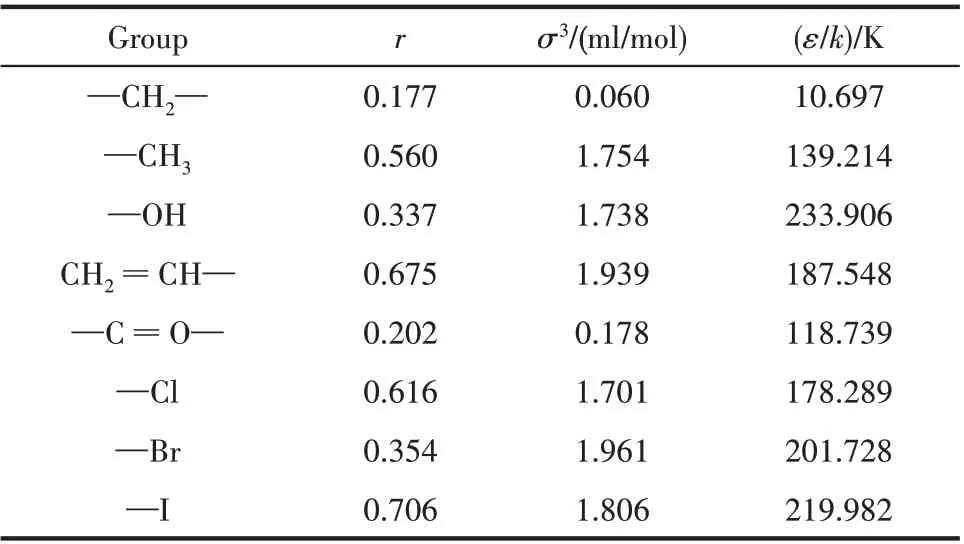
表1 基团对模型参数的贡献值Table 1 Group contribution value to model parameters
获得基团参数后,即可利用式(2)和式(3)确定不同物质的模型参数,再利用状态方程式(1)即可确定pVT关系了。对于低碳直链烷烃C3~C9的密度,GCSWCF 预测的总的平均误差为1.73%。对于高碳烷烃(C10~C18),密度预测总的平均误差为2.39%。说明GC-SWCF 能在较宽的温度范围内较好地预测直链烷烃的密度。对于同样的系统,Tamouza 等[24]采用GC-SAFT-0 和GC-SAFT-VR 预测时误差分别为3.93%和3.97%;Mattedi 等[25]采用GC-LF 的误差为2.87%。应该指出,对于直链烷烃,模型只涉及两类基团,即—CH2—和—CH3,它们对分子参数的贡献依据的是C3~C9的实验数据,但将其拓展到碳数≥10的直链烷烃,得到的效果依然满意,说明GC-SWCF建立的思路是正确的。


图1 正丙烷-正癸烷混合物密度的计算结果(实线)和实验值(点)[26]的比较Fig.1 Comparison of calculated(lines)and experimental(points)[26]densities for n-propane and n-decane mixture
图2 示意了烷烃(C5~C8)-醇二元系密度的GCSWCF 方程计算结果与实验值[27]的比较,误差分别为1.41%、1.53%、1.31%和0.39%。应该指出,由于醇分子间存在氢键缔合,但GC-SWCF 方程并未考虑缔合作用,因而缔合的影响将被分布到其他基团之中。在同样未考虑缔合的影响下,GC-SAFT-0的计算结果分别为0.86%、1.09%、2.66%和2.38%[13],GC-SAFT-VR 则 分 别 为0.89%、1.14%、2.47% 和1.40%[13]。比较而言,GC-SWCF 的计算结果还是令人满意的。
图3 给出了己烷-乙醇二元系汽液相平衡GCSWCF 的计算结果(实线)和实验值(点)[28]的比较。计算时采用的可调参数kkl= 0.05355。由图可见,在己烷含量较低时,GC-SWCF 计算值稍比实验值大。随着混合物中己烷摩尔分数的增大,GC-SWCF 的结果将完美趋近实验值。总体而言,恒压下,计算的温度误差为1.36 K,气相组成误差为3.67%。恒温下,计算的压力误差为6.72%,气相组成误差为3.79%。

图2 烷烃-醇混合物密度的计算结果(实线)和实验值(点)[27]的比较Fig.2 Comparison of calculated(lines)and experimental(points)[27]densities for alkane and alcohol mixture

图3 乙醇-己烷二元汽液相平衡的计算结果(实线)和实验值(点)[28]的比较Fig.3 Comparison of calculated(lines)and experimental(points)[28]VLE for ethanol and 1-hexane binary system
表2 列出了部分二元系汽液平衡的关联结果,实验数据取自文献[28]。其中,温度平均偏差为1.84 K,压力平均偏差为3.20 kPa,气相组成平均相对偏差为1.98%。采用GC-SAFT-0 时,压力的平均偏差为4.31 kPa,气相组成的平均相对偏差为2.02%[13]。采用GC-SAFT-VR 则压力的平均偏差为6.10 kPa,气相组成的平均相对偏差为2.89%[13]。可见,采用与温度无关的可调参数后GC-SWCF 可满意应用于混合物汽液相平衡的计算中。表2也给出了GC-SWCF 的预测结果,此时kkl= 0。可见,对于分子结构与性质相近的物质所组成的混合物,GCSWCF的预测结果也可接受。
应该指出,按上述思路建立的基团贡献状态方程拓展到混合物时,必须使用可调参数计算不同链节间的交叉作用能。为克服这一弊端,一是直接建立“基团基”的基团贡献状态方程,方法是将模型中原先的“链节”改用“基团组合”替代。由于“基团组合”遍及系统中所有物质的不同基团,实际应用时并不需要可调参数,如GC-SAFT-VR[29-30]、GCSAFT-γ[31],以及基团贡献格子流体状态方程(GCNLF)[32]等。二是采用London 理论确立可调参数,方法是利用链节的虚拟离子化能量确定可调参数[15,33-34]。当采用基团贡献计算链节虚拟离子化能量时,可调参数也可实现基团贡献化[35]。三是结合基团贡献活度系数模型建立状态方程[36-38],它们实际上是不同理论的组合,应用时还需要物质的临界参数或偏心因子。

表2 二元系汽液相平衡关联结果Table 2 Correlated results of VLE for binary system
2 热力学模型参数的理论预测
显然,分子热力学模型的核心除了所能表达的物理意义之外,就是其自身的参数。一套完备的化合物的模型参数是任何一个分子热力学模型广泛使用的必要条件。化工设计软件都因其具有强大的物性数据库和参数数据库而受到广泛应用。这些软件的数据库都来自于数据手册和海量的文献资料,它们是大量实验结果的积累。同样,基团贡献法中基团参数的确定也离不开可靠且具代表性的实验数据。当实验数据缺失时,分子热力学模型参数将无法确定。因此,探索一种不依赖实验数据而确定分子热力学模型参数的方法就显得至关重要了。
对于立方型状态方程,理论上可根据物质的临界性质(少数也需要偏心因子)确定模型参数,尽管也发展了不同基团贡献法预测临界性质或偏心因子,但目前的基团贡献法仍不能区分同分异构体,特别是分子中存在不同官能团时,基团贡献法也不能全面反映官能团间的相关性,这都限制了模型的有效应用。可见,理论预测模型参数的终极目标应该是直接通过分子的结构确定参数。
不可否认,构成分子的官能团一定,其在空间排布的方式一定,则模型参数也应一定。2008 年,一种基于abinitio溶剂化计算确定立方型状态方程参数的理论方法已被提出[39]。在这一方法中,状态方程中相互作用参数和体积参数可根据理论计算的溶剂化自由能和溶剂化空穴体积确定。所有计算均可采用不同版本的COSMO(conductor-like screening model 似导体屏蔽模型)完成,首先是量子化学软件计算获得COSMO 量化数据,其次是统计力学计算以获得溶剂化自由能,需要输入的仅是元素的特定参数和一些普适性参数。Panayiotou[40]建立了COSMO 理论与非随机氢键模型(NRHB)间的关系,提出了格子模型中参数的COSMO 计算方法。由于COSMO 结合了量子力学与统计力学,因此,基于COSMO 计算模型参数完全是一种不依赖实验数据的理论预测方法,还可推广到混合物而不必引入可调参数[41]。此外,也可采用分子动力学模拟方法确定格子模型中的能量参数[42-43],通常采用商业软件的专用模块计算,但结果需校正。
COSMO 计算的关键是分子的构型优化,虽然耗时,但结果能体现分子的构象和同分异构体。因此,基于量化计算所确定的分子热力学模型参数也与结构一一对应,弥补了基团贡献法的不足。尽管不同版本的COSMO 方法都可直接用于流体混合物相平衡计算,但方法本身需要纯物质在不同温度下的饱和蒸汽压数据。若一旦基于COSMO 模型确定热力学模型中的参数后,则能实现蒸汽压、汽化焓、密度和混合物相平衡计算[39-40],进一步则能考察流体的界面性质和结构。当热力学模型嵌入化工模拟软件中,则可实时在线对过程进行模拟、计算与分析。很遗憾,目前基于COSMO 方法还仅限于两参数热力学模型,尝试将这一方法拓展到SWCFVR 方程中,借以从分子结构出发预测SWCF-VR 中的模型参数,可称为COSMO-SWCF。
前已述及,非缔合型SWCF-VR 状态方程包含4个模型参数,链节数r、链节直径σ、方阱阱深ε和方阱宽度λ。为简化,固定方阱阱宽λ=1.5。链节数r和链节直径σ通过COSMO 获得的空穴体积Vcosmo和空穴面积Acosmo确定。它们间对比关系如式(4)所示
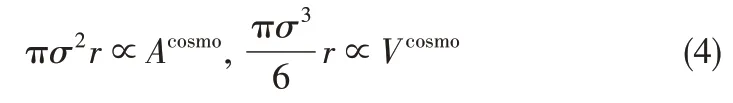
方阱阱深可通过溶剂化自由能ΔG∗sol与状态方程的关系确定,关系式如式(5)[39,44-45]所示
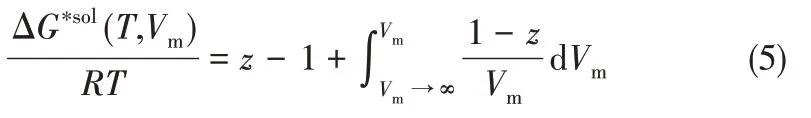
式中,Vm为系统的摩尔体积,m3;T为系统温度,K;R为气体普适性常数,J/(mol·K)。
基于COSMO 确定SWCF-VR 模型参数的基本步骤是,首先画出物质的分子结构并优化结果,接着利用COSMO 原理获得的空穴体积Vcosmo和空穴面积Acosmo依据式(4)确定分子的链节数和链节直径,并通过溶剂化自由能的COSMO 预测结果与SWCFVR 的计算结果的对应关系采用式(5)确定模型参数中的方阱阱深。通过此方法,已获得了烃类、卤代烃类及含氧有机物(醇、酯、醚、酮)等十大类共192种有机化合物的模型参数[46],发现基于COSMO 获得的模型参数与直接采用实验数据获得的参数都能反映分子结构的变化规律。
利用得到的模型参数即可计算物质的pVT关系与相平衡数据了。图4示出了不同温度下直链烷烃密度的计算结果与实验数据[47]的比较,其总的平均相对误差为0.23%。Voutsas 等[48]分别运用PR 方程、PR-fitted (PR-f)方程、Sanchez-Lacombe (SL)方程、SAFT 方程、PC-SAFT 方程对直链烷烃密度进行了计算,其总的平均相对误差分别为2.8%、3.5%、3.6%、5.5%和2.1%。可见,结合COSMO 方法得到的SWCF-VR 模型参数应用于直链烷烃密度计算时,获得了令人满意的结果。
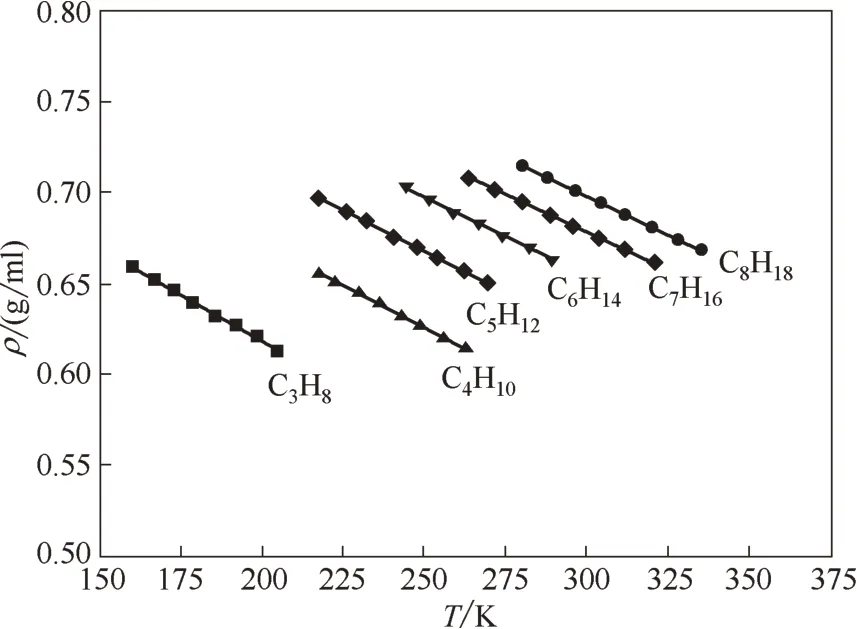
图4 直链烷烃密度的计算结果与实验值[47]的比较Fig.4 Comparison of calculated(lines)and experimental(points)[47]densities for n-alkanes
采用COSMO 只能获得纯物质的模型参数,当SWCF-VR 应用于混合物时,仍需要利用少量实验数据拟合二元可调参数kkl以获得分子k和分子l间的交叉作用参数εkl。图5 示出了313.15 K 下烷烃(1)-苯(2)二元混合物密度的模型计算值与实验值[49]比较。其中,己烷、庚烷、辛烷、壬烷分别与苯组成的混合物,其密度计算的平均相对误差分别为0.52%、0.51%、0.55%和0.48%。可见,采用一个与温度无关的可调参数,模型同样能应用于二元混合物的密度计算。
表3 列出了部分二元系汽气液平衡的计算结果,实验数据取自文献[28]。由表可见,当系统含醇等缔合物质时,SWCF-VR 方程关联误差较大。如不存在氢键缔合作用时,关联效果会得到明显改善,这是由于目前基于COSMO 确定模型参数时并未考虑缔合作用。总体而言,模型关联的汽相组成总的平均相对偏差为3.08%。
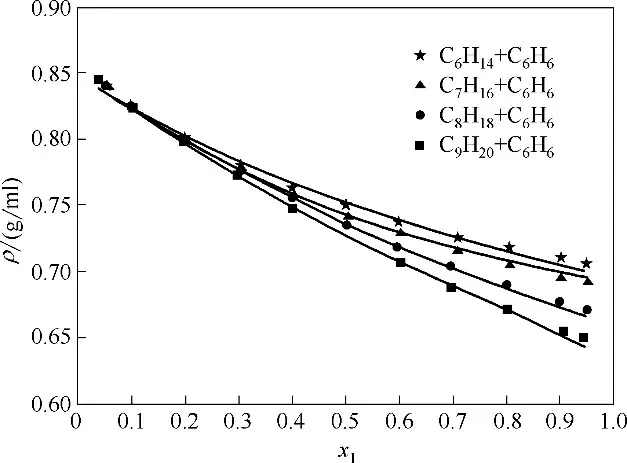
图5 烷烃(1)-苯(2)混合物密度计算值与实验值[49]的比较Fig.5 Comparison of calculated(lines)and experimental(points)[49]densities for alkanes(1)+benzene(2)
显然,目前的工作仅停留在基于COSMO 确定纯物质的SWCF-VR 参数,当模型应用于混合物时,必须使用可调参数确定不同链节间的交叉作用能。理论上,也可根据混合物的溶剂化自由能通过式(5)的方式确定不同链节间的交叉作用能。
3 结 论
分子热力学模型的有效应用离不开模型参数的确定。为赋予SWCF-VR 预测功能,一是建立基团贡献状态方程GC-SWCF,已获得了不同基团对模型参数的贡献值。结果证明GC-SWCF 方程能满意预测纯物质的密度,并能在相对宽的温度与组成范围计算二元混合物的密度。二是基于COSMO 模型从物质结构出发直接得到模型参数,这是一种完全不依赖实验数据确定模型参数的理论方法。本文已基于COSMO 获得了烷烃类、烯烃类、炔烃类、环烷烃、芳香烃、卤代烃、酮、醚、酯、醇等十类共192种有机化合物的模型参数,发现COSMO+SWCF 能满意预测纯物质的密度,也能满意计算二元混合物的密度。引入一个与温度无关的可调参数后,GCSWCF 与COSMO+SWCF 都可应用于汽液相平衡的计算中,但对缔合系统效果稍差。SWCF 方程虽然是为了研究流体pVT关系和相平衡而建立起来的,但与其他理论结合也可构建黏度模型和界面张力模型。这说明,GC-SWCF 以及COSMO+SWCF 也可推广应用于流体黏度和界面张力的计算中,但本质依然是SWCF方程中模型参数的确定问题。应该指出,目前理论确定模型参数的研究还仅限于常规物质,尚未推广到离子液体系统。近些年来,各种功能基团修饰的离子液体不断涌现,基于COSMO 模型筛选离子液体已初见成效,但系统的pVT关系和相平衡性质研究则主要还是靠热力学模型。如能建立二者间的联系,采用热力学模型预先确定热力学性质则能起到事半功倍的效果。可见,基于COSMO+SWCF 方法对于指导设计多功能离子液体具有重要意义。

表3 二元系汽液相平衡关联结果Table 3 Correlated results of VLE for binary system
符 号 说 明
Acosmo——空穴面积,m2
G*sol——溶剂化自由能,J
N——分子数
p——压强,MPa
R——气体普适性参数,8.3145 J/(mol·K)
r——链节数
T——系统温度,K
Vcosmo——空穴体积,m3
Vm——系统摩尔体积,m3
x——摩尔分数
y——气相组成摩尔分数
z——压缩因子
ε——阱深,J
λ——阱宽
ρ——密度,g/ml
σ——链节直径
上角标
cal——计算值
exp——实验值
HS-chain——硬球成链的贡献
HS-mono——硬球单体对压缩因子的贡献
SW——方阱
SW-mono——方阱单体的贡献
Δ(HS-Chain)——方阱作用对成链的影响
下角标
i——组分
