HPLC法测定孕穗期水稻剑叶中多种内源激素的含量
2020-03-05尚蓉霞余欣石鸿宇吴汉武立权李娟
尚蓉霞余欣石鸿宇吴汉武立权*李娟
(1安徽农业大学农学院,合肥230036;2安徽农业大学生命科学学院,合肥230036;3安徽省淠史杭灌区灌溉试验总站,安徽六安 237158;4安徽农业大学生物技术中心,合肥 230036;第一作者:18235445708@163.cm;*通讯作者:Wlq-001@163.com;lj_welcome_ok@126.com)
植物内源激素是指由植物自身合成的一类生物活性物质。它们只以微小计量(含量一般为组织鲜质量的10-9~10-7数量级)即可对植物的生长发育、细胞分化、开花结果、抗逆性以及器官衰老过程进行调控[1-2]。在正常环境条件下,植物在各种激素相对平衡的调节下进行正常的生长发育和新陈代谢;但当遭遇逆境时,激素间的平衡遭到破坏,就会使植物的生长发育紊乱、新陈代谢失调[3]。
由于植物内源激素含量极低,性质不稳定、易分解,且前处理过程不能完全消除植物组织中复杂的基质干扰,因此获得一种灵敏度高、专一性强、操作简便、耗时短的检测方法一直是国内外诸多学者研究的热点[4]。传统的检测方法有生物检测法、免疫检测法和基于光谱或色谱的理化分析法。生物鉴定法最早用于检测植物激素,它利用当激素作用于植物组织与器官时会产生特异性反应这一原理实现对植物激素的测定。生物鉴定法虽然方法简便,但对样品纯度要求较高,需要复杂的样品前处理作为支撑,同时由于其专一性、重复性较差,这类方法的应用逐渐减少[5]。免疫检测法中以酶联免疫吸附测定法(ELISA)最为常用,ELISA法需购买不同的试剂盒,这就使得被测组分和同一激素不同组分的定量分析被限制,且各组分之间存在交叉反应,降低了其灵敏度和选择性,从而影响测定结果的准确性[6]。最早的光谱法是用比色法测定IAA,但是灵敏度和专一性较低,目前基本不再使用该方法。后来开发了红外吸收光谱法、紫外吸收光谱法及荧光法等,虽然检测的灵敏度得以提升,但其专一性较差,因而往往应用于对激素分子的结构鉴定及定性分析,定量分析较少采用该类方法[7]。现代分析植物激素最常见的色谱技术主要包括液质联用(LC-MS)、气相色谱法(GC)和高效液相色谱法(HPLC)。LC-MC费用颇高;GC法灵敏度高,但不同类激素要求不同的前处理和衍生化方法,难以同时测定多个激素,进而增加了材料的用量[8];HPLC是一种高效分离纯化技术,具有灵敏度、分辨率都高,专一性强和重现性好等优点,被广泛用于植物激素测定(除乙烯外)[9-11]。
水稻是我国的主要粮食作物。在水稻生产过程中,其产量和品质常常会遭受高(低)温、干旱、盐渍等非生物逆境的影响[12-13]。了解水稻内源激素在应对不同逆境胁迫中的调节规律,可为其抗逆栽培提供重要的生产实践价值。但测定水稻中5种内源激素方法的研究很少。因此,笔者建立HPLC法来检测水稻中的5种内源激素。
1 实验方法
1.1 仪器与试剂
Waters600高效液相色谱系统(美国Waters公司);超纯水制备系统(美国Millipore公司);JW-3021HR高速冷冻离心机(嘉文);ZHWY-200B恒温培养振荡器;Centrifuge 5417R超速冷冻离心机(eppendorf);MGC-300H人工气候箱。ProElut SPE C18200 mg/3 mL。C185 um×4.6×250 nm。
玉米素(Z)(纯度>98%,aladdin)、3-吲哚乙酸(IAA)、水杨酸(SA)(纯度>99%,aladdin)、赤霉素(GA)和反式脱落酸(ABA)(纯度分别≥96%和>98%,MACKLIN)、甲醇和乙酸(色谱纯,美国TEDIA公司)、石油醚(分析纯),其他试剂均为分析纯,实验用水均为超纯水(18.2 MΩ·cm)。所有试剂在使用前均用 0.22 μm 有机膜过滤。
1.2 标准溶液配制
精确称取Z、GA、IAA、ABA和SA标准品分别为0.2 mg、2 mg、0.2 mg、0.1 mg 和 0.4 mg 于 2 mL 的容量瓶,用甲醇溶解并定容至刻度,配制浓度分别为0.1 mg/mL Z、1 mg/mL GA、0.1 mg/mL IAA、0.05 mg/mL ABA和0.2 mg/mL SA的标准储备溶液。用甲醇将储备液依次稀释2倍,连续进行4次,配成5份浓度依次相差2倍的系列标准溶液,以实际注入标准物的量为横坐标,相应的峰面积为纵坐标作图。
1.3 样品前处理
试验于2017年5月26日在安徽农业大学生物科技楼人工气候室进行,以籼稻品种Nagina 22(Oryza sativaaus)为试验材料。选种、消毒、浸种和催芽,选取发芽均匀一致的籽粒种于带网格的塑料盆(高30 cm,内径25 cm)中,每盆3株,一共72盆,置于人工气候室培养;盆内盛有国际水稻Hoagland营养液(霍格兰配方)[14],每3 d更换1次营养液;气候室生长条件为昼/夜温度为30℃/25℃,昼/夜光周期为12 h/12 h,相对湿度为75%,光照强度为3 000 Lx。本试验各处理除温度和模拟干旱外,其余环境设置均相同。试验设正常生长(CK)、单一干旱(DT)、单一高温(HT)及干旱–高温同时(DH)4个处理。以叶龄余数法[15]诊断幼穗发育进程,此时大多数稻穗进入花粉母细胞减数分裂期[16-17]。其中,(1)正常对照:Hoagland营养液中不添加10% PEG-6000,昼夜温度为 30℃/25℃;(2)单一干旱处理:在已有文献[18]和前期预备试验基础上,明确了10% PEG-6000可使水稻达到中度干旱胁迫程度,故本研究确定采用10% PEG-6000 Hoagland浇灌模拟中度干旱胁迫,每处理重复3次,每重复6盆,培养3 d;(3)单一高温处理:除人工气候室昼夜温度变为38℃/25℃外,其它条件不变[19],高温连续处理3 d,每处理重复3次,每重复6盆;(4)干旱-高温同时处理:用10% PEG-6000 Hoagland浇灌,且设高温38℃/25℃处理3 d,每处理重复3次,每重复6盆。胁迫结束后,取水稻剑叶用超纯水洗净、擦干。准确称取水稻剑叶样品1g放入研钵,用液氮研磨,再转移到5 mL的离心管中,加入3 mL 80%的冷甲醇,置于4℃冰箱浸提过夜。加1 g的 PVPP,以 10℃震荡 20 min,之后以 12 000 rpm、4℃离心10 min,取上清于新离心管。向残杂中加入1 mL 80%的冷甲醇,重复上一步,取上清合并,并过SPE C18柱(先分别用甲醇和70%的甲醇各5 mL活化)去杂纯化,再用0.22 μm的微孔有机滤膜过滤,然后收集滤液,-20℃保存备用。用过的SPE C18柱可以采用纯甲醇和乙醚洗去亲酯色素,以便重复利用。
1.4 色谱条件
色谱柱:C18反相色谱柱(5 μm×4.6×250 nm);流动相A为甲醇,C为0.5%乙酸的水溶液;流速为0.9 mL/min;进样量为20 μL;紫外检测波长设置为272 nm、236 nm和254 nm以及全扫波段;柱温为30℃。洗脱梯度见表1。
2 结果与分析
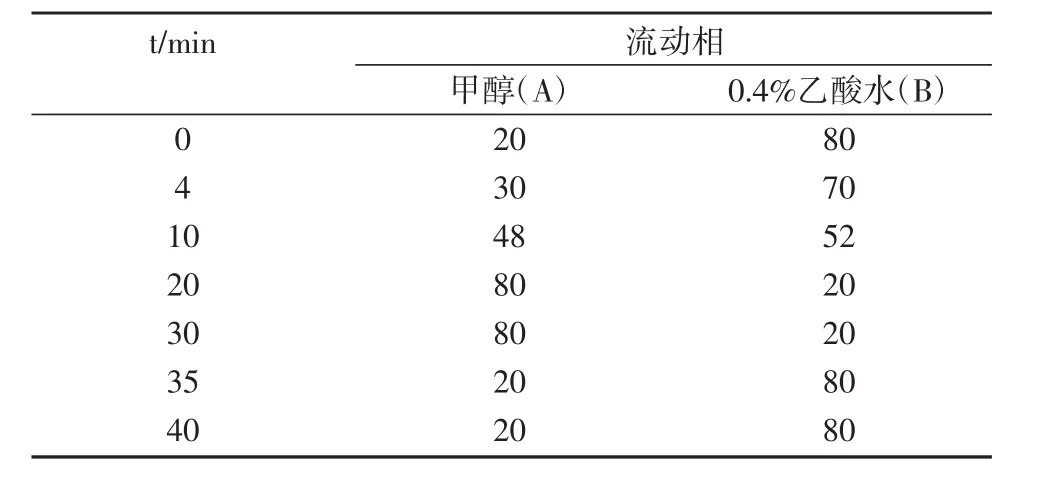
表1 流动相的洗脱梯度比例 (%)
2.1 色谱条件的优化2.1.1 流动相的选择
分别采用甲醇-水和甲醇-乙酸水作为流动相进行分析测定。结果表明,以甲醇-水为流动相时,色谱峰分离效果不是很好;以甲醇-乙酸水为流动相时5种标品能得到很好的分离,且色谱峰的峰形尖锐、理想。因此,选择甲醇-乙酸水为流动相。配制了体积分数分别为0.1%、0.2%、0.3%、0.4%和 0.5%的乙酸水溶液。色谱峰分离结果表明,当乙酸水溶液为0.5%时,5种激素可获得较好的分离效果和色谱峰形。
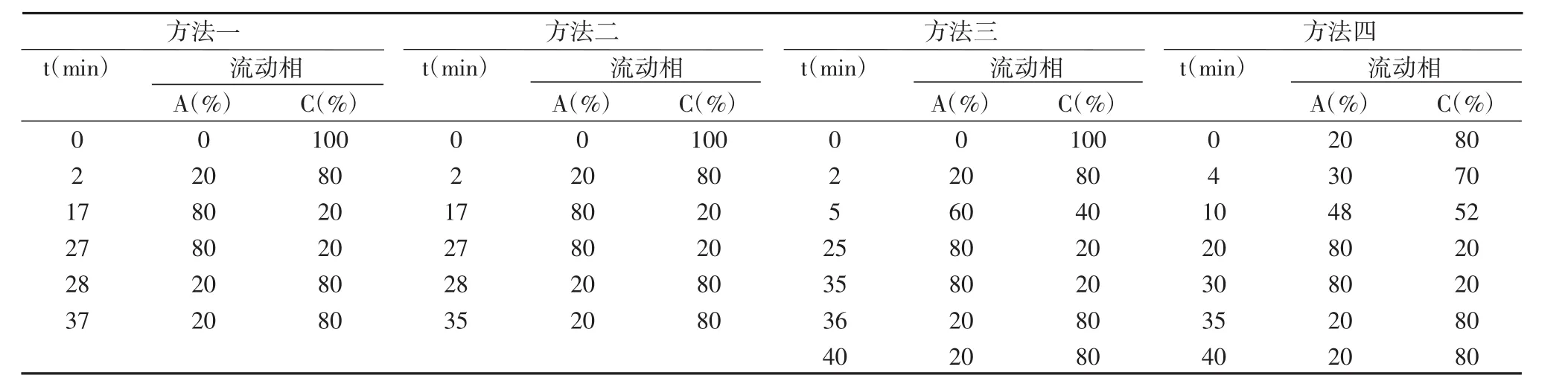
表2 植物内源激素洗脱条件
2.1.2 流动相pH值、配比的选择
用乙酸分别调节流动相 pH 值为 2.5、3.0、3.5。结果发现,流动相的pH值变化直接影响化合物的分离效果,当流动相pH值为3.0时,各化合物获得最佳分离效果。因此选择pH值为3.0的乙酸水溶液。
本实验考察了流动相甲醇与水的比例对5种标准品激素的洗脱速度和分离度的影响。流动相洗脱时间与比例如表2所示,与方法一、二和三相比,方法四使得5种标准品的分离效果较好。因此,我们选择方法四。
2.1.3 吸收波长的选择
我们设置5个波长,分别是236 nm、240 nm、246 nm、254 nm和272 nm,发现水杨酸在236 nm时波形尖锐、理想;玉米素、生长素和脱落酸在波长为272 nm时,峰型效果好;但由于赤霉素含量很低,因此,我们选择赤霉素的最大吸收波长,为254 nm。而波长为240 nm和246 nm不能满足峰型尖锐。因此,我们将本试验的检测波长设为为236 nm、254 nm和272 nm。5种激素的标准品的色谱图如图1所示。
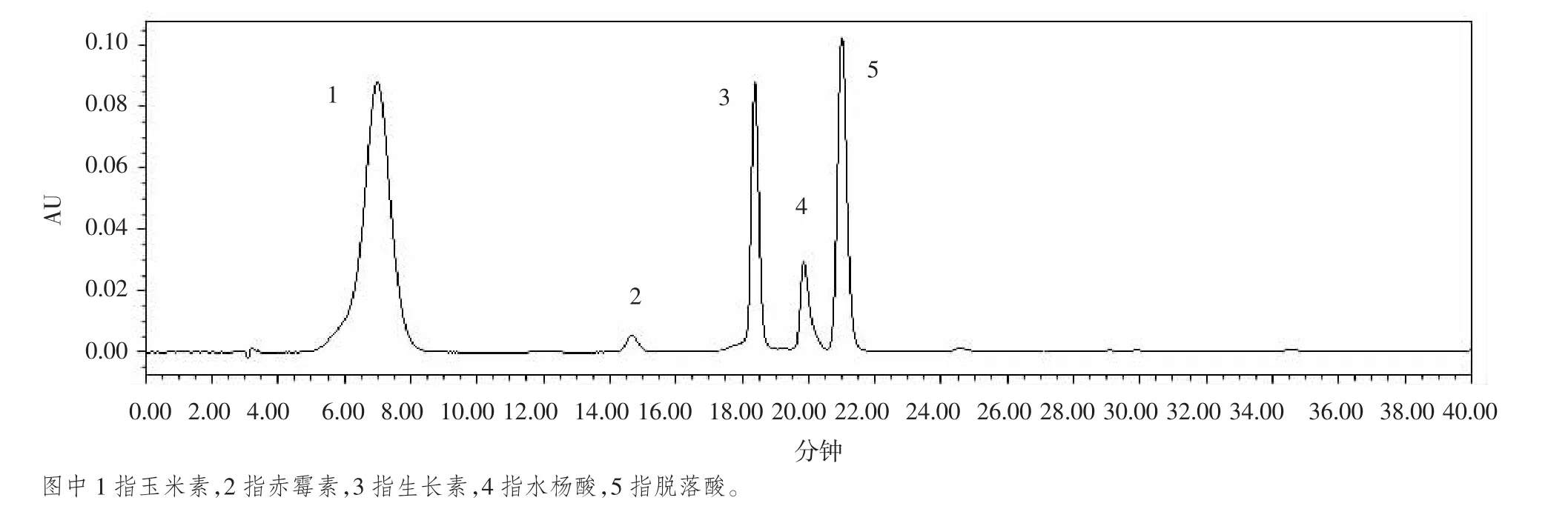
图1 五种植物内源激素标准品的色谱图
2.2 标准曲线及其线性范围
标准品质量浓度范围在3.125~1 000 mg/kg之间,设置5个梯度,在上述色谱条件下进行检测,以质量浓度(x)与峰面积(y)进行线性回归,得出各激素标准品的标准曲线(见表 2),相关系数 R2>0.9996,以 3 倍信噪比(S/N=3)求得5种激素的检出限,并以线性范围的下限浓度作为本试验实测样品的定量限。结果表明,5种激素标准品在3.125~1 000 mg/kg范围内线性良好,可满足定量分析的要求。混合标准品出峰如图1。
2.3 回收率和精密度试验
为减少实验过程的人为污染,确保实验的准确性,需要对水稻剑叶样品进行加标回收率的测定。称取已知含量的样品3份,精密加入Z、GA、IAA、SA和ABA标准储备溶液高浓度、中浓度和低浓度3个浓度1 mL,按实验方法进行操作测定,每个样做3次平行实验,根据加入标准品的质量浓度与检出质量浓度计算回收率和相对标准偏差(RSD),结果见表4。5种激素的回收率在 45.73%~116.70%之间,RSD 范围为 0.75%~26.72%,回收效果较好,精密度较高。
2.4 水稻剑叶样品的测定
按1.3样品提取、纯化方法处理样品,利用建立的方法对高温、干旱及高温干旱同时胁迫处理的水稻剑叶内源激素的含量进行测定。结果(表5)显示,与CK相比,HT、DT和DH处理的水稻剑叶Z、GA和IAA含量都下降,而SA和ABA含量明显上升。胁迫处理下,Z的含量比 CK 分别低 8.63%、35.08%和 39.27%;GA 的含量比 CK 分别低 40.85%、34.76%和 76.22%;IAA 的含量比 CK 分别低 38.62%、21.26%和 43.78%;SA 的含量比 CK 分别高 16.54%、25.78%和 18.28%;ABA 的含量比 CK 分别高 30.00%、48.75%和 171.25%。DH 处理下,Z、IAA、GA和SA含量基本上低于DT和HT处理,而ABA含量明显高于DT和HT处理。
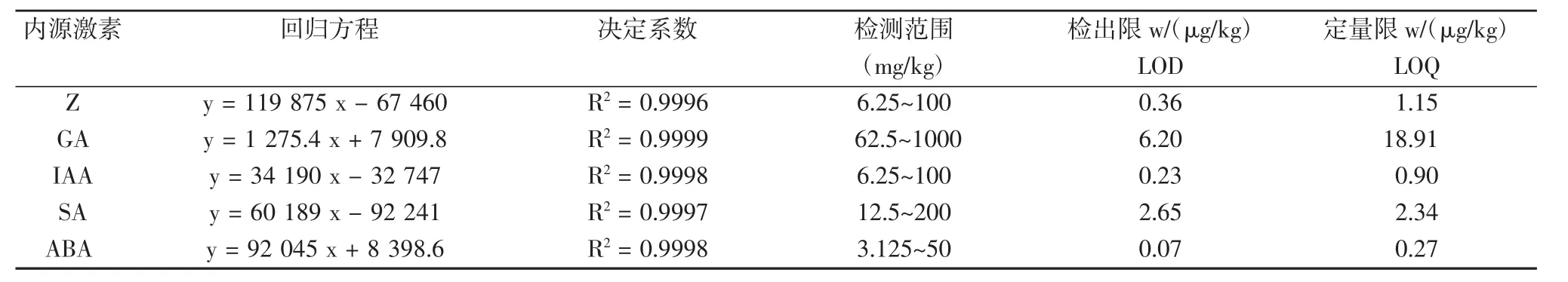
表3 回归方程、相关系数、检测范围、检出限和定量限
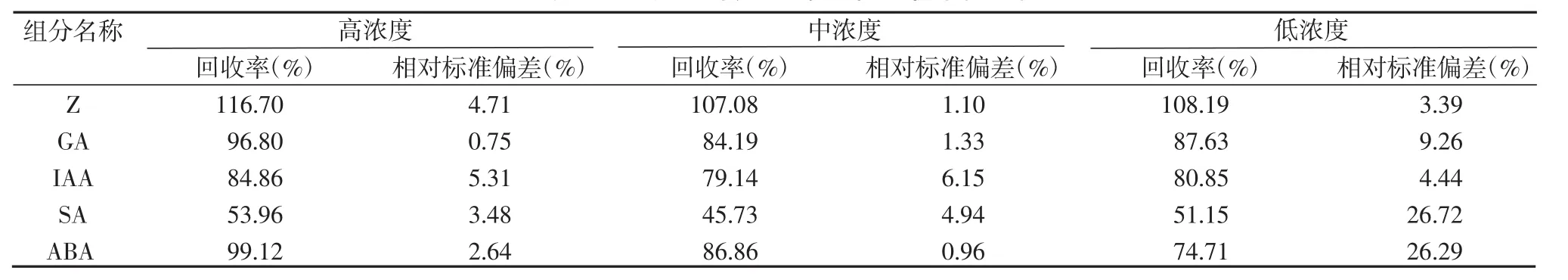
表4 五种植物内源激素加标回收率(n=3)

表5 不同胁迫处理下孕穗期水稻剑叶各内源激素的含量 (μg/g)
3 讨论
本文通过高效液相色谱法快速、有效地检测不同胁迫处理孕穗期水稻剑叶内源激素的含量。峰形尖锐、分离效果理想,加标回收率达 45.73%~116.70%,方法能满足内源激素定量测定的要求,为进一步检测水稻生长过程中内源激素的变化与水稻生长、衰老等相关性研究提供了关键技术。运用此方法测定高温、干旱以及高温干旱同时胁迫孕穗期水稻剑叶内源激素的含量,结果表明,Z、IAA和GA的含量胁迫处理要比正常生长的低,且复合胁迫处理比单独胁迫的还要低,说明复合胁迫增强了对小穗的伤害。
