细胞焦亡研究进展①
2020-02-20黄清宇杜楚江张雨竹曹宏伟郝慧芳
黄清宇 杜楚江 张雨竹 曹宏伟 郝慧芳
(内蒙古大学生命科学学院,呼和浩特 010021)
细胞死亡在机体发育、维持内环境稳态和疾病的发生发展过程中起关键作用,人们对细胞死亡的认识经历了一个漫长的过程,随着研究的不断深入细化,基于不同的划分标准,细胞死亡也有多种不同的种类。程序性细胞死亡方式(programmed cell death,PCD)可以分为裂解性细胞死亡和非裂解性细胞死亡[1], 细胞凋亡(apoptosis)是非裂解性死亡的形式,不会引起炎症反应的发生[2]。坏死性凋亡(necroptosis)和细胞焦亡(pyroptosis)属于裂解性细胞死亡的类型,这两种死亡使得细胞内含物外漏,引起炎症反应,因此称为炎症性死亡[3,4]。除此之外,关于细胞死亡的研究还在不断地进行,如近些年研究发现的新型细胞坏死方式——铁死亡(ferroptosis)[5]。研究细胞死亡的机制可为一些疾病的基础研究和临床治疗提供新的思路。
细胞焦亡是最近几年新发现的一种细胞程序性死亡的方式,是机体中主要的非特异性防御机制,在拮抗外部病原体入侵和感知内源危险信号中发挥着不可替代的作用。本文就近年来细胞焦亡的发现和命名、形态学和分子特征、分子机制和焦亡相关的疾病进行综述。
1 细胞焦亡的发现和命名
早在1992年,Zychlinsky等[6]在实验中观察到弗氏志贺菌可诱导感染的宿主巨噬细胞发生裂解性死亡。这种裂解性死亡的形式具有细胞凋亡的部分特征,例如染色质凝集、DNA断裂及有半胱氨酸蛋白酶(Caspase)活性依赖性,因此,最初这种死亡方式被认为是凋亡。2001年,华盛顿大学学者Cookson等[7]研究表明,该死亡形式具有Caspase-1活性依赖性,而不同于Caspase-3活性依赖的细胞凋亡。他们首次将细胞焦亡定义为Caspase-1依赖的细胞死亡的形式。“pyro”是火的意思,表明这种程序性细胞死亡引起炎症反应,而“ptosis”是落下的意思,表明其程序性死亡的本质。2014,2015年邵峰院士研究团队发现细胞焦亡还可以由胞质内LPS对Caspase-4/5/11激活所引起,活化的Caspase-4/5/11通过对Gasdermin家族蛋白的切割最终诱发细胞焦亡。因此,他们把细胞焦亡定义为Gasdermin家族介导的细胞程序性坏死[8,9]。
2 细胞焦亡的形态学和分子特征
2.1细胞凋亡与细胞焦亡 长期以来,人们一直认为细胞凋亡是唯一的细胞程序性死亡方式,对其形态学特征及分子机制已经有了一个较深入、完善的了解。细胞凋亡起始表现为细胞表面的微绒毛等特化结构逐渐消失,细胞皱缩并与周围细胞分离,胞质和核染色质固缩,在细胞内特异性核酸内切酶作用下,核染色质断裂降解为约180~200 bp或其整数倍核苷酸小片段,直到后期凋亡细胞的内容物包裹细胞质膜形成凋亡小体,被附近的间质细胞或巨噬细胞吞噬。在此期间细胞膜保持一定的完整性,故不引发炎症反应。将凋亡细胞中的DNA提取出来经过琼脂糖凝胶电泳,会呈现出梯状条带,即DNA ladders,用转移酶介导的dUTP缺口末端标记测定法(TdT-mediated dUTP nick-end labeling,TUNEL)和对翻转到细胞外侧的磷脂酰丝氨酸敏感的Annexin V检测,呈现出染色阳性[10,11],如表1所示。
细胞焦亡在形态学上兼具细胞坏死和凋亡的部分特点,细胞发生焦亡时,细胞核浓缩、染色质DNA随机断裂降解,细胞膜上出现众多孔隙,细胞膜失去调控物质进出的能力,细胞丧失内外离子平衡,发生渗透性肿胀进而膜破裂,释放出细胞内容物等活性物质,激发机体的免疫反应,募集更多的炎症细胞,并扩大了炎症反应。经过TUNEL和Annexin V染色呈现阳性[1,3],如表1所示。
2.2细胞焦亡的分子特征
2.2.1Caspase家族 半胱天冬酶(Caspase)家族是细胞质中具同源性、相似结构特征的蛋白水解酶,可在下游靶蛋白天冬氨酸残基后的肽键进行有选择地识别切割。在正常的细胞中,Caspase蛋白通常都是以没有活性的酶原状态(Pro-caspase)存在,氨基酸序列水解后变为有活性的Caspase才能发挥其作用。至今为止,在哺乳动物中已经得到证实有15种Caspase家族成员,在人类中已经发现了13种Caspase,在小鼠中有11种Caspase[12]。根据结构和功能的差异,Caspase可以分成凋亡类和炎性类两种。其中凋亡Caspase包括Caspase-2/3/6/7/8/9/10,以Caspase-3为代表,与细胞凋亡有关;炎性 Caspase包括Caspase-1/4/5/11/12/13/14,介导炎症反应[13,14]。炎性Caspase-1和Caspase-4/5/11的激活最终引发了细胞焦亡的发生,其发生的具体分子机制将在下一部分详细讲述。
2.2.2Gasdermin家族 Gasdermins是功能多样的蛋白质家族,在多种细胞类型和组织中表达,人类Gasdermins由Gasdermin A(GSDMA)、Gasdermin B(GSDMB)、Gasdermin C(GSDMC)、Gasdermin D(GSDMD)、Gasdermin E(GSDME,又称DFNA5)和Pejvakin(PJVK,又称DFNB59)组成[15]。除PJVK外,所有Gasdermins均有保守的双结构域排列:C-末端结构域(GSDM-C)和N-末端结构域(GSDM-N),N末端具有成孔活性,具有诱导细胞发生焦亡的特征[16,17]。
GSDMA、GSDMB主要在食道、肠道细胞中表达,与脱发、哮喘及炎症疾病等相关。人类GSDMC蛋白在胃、食管、脾的上皮细胞中表达,在胃癌等癌症细胞中被抑制,生物学功能尚在研究[18]。GSDMD、GSDME广泛表达于不同细胞组织,GSDMD可被炎性Caspase-1、4、5、11特异性激活,切割成GSDMD-N(p30片段)和GSDMD-C(p20片段),GSDMD-C存在于胞质中,GSDMD-N具有亲脂特性,与细胞膜内侧的磷脂酰肌醇、细菌质膜外侧的心磷脂特异性结合,在膜中寡聚化并形成直径为10~16 nm的孔[19],通过该孔分泌较小直径的底物,最终导致膜破裂,释放出整个细胞内容物[20]。GSDME在受到化疗药物、肿瘤坏死因子(tumor necrosis factor,TNF)和病毒感染刺激时,可被凋亡信号通路的Caspase-3活化,对细胞膜打孔,将本应发生凋亡的细胞转化为焦亡[21,22]。通常GSDME会在正常细胞中高水平表达,而癌细胞经过DNA甲基化和组蛋白等表观遗传修饰[23],多数处于GSDME抑制表达或低水平表达状态,在“凋亡刺激”下,高表达GSDME的正常细胞发生焦亡可能是常规化疗药物产生毒副作用的原因之一。
表1 凋亡和焦亡的特征
Tab.1 Features of apoptosis and pyroptosis

FeaturesApoptosisPyroptosisCell morphological featuresThe cells shrink and become smallerThe cells develop osmotic swelling, become larger, and eventual lysisChanges in plasma membraneThe plasma membrane remained largely intact until it became apoptotic bodiesThe integrity of the membrane is lost, and pores form in the plasma membraneChanges in chromosomesChromatin DNA is cleaved to approximately 180-200 bp or its integer nucleotide fragmentsRandom fragmentation of chromatin DNA occursInflammatory reactionThere is no intracellular content release and no in-flammatory responseThe proinflammatory intracellular contents release and cause a cascade of amplified inflammatory re-sponses
3 细胞焦亡的机制研究
当微生物外源或内源性感染宿主细胞时,位于胞质内的模式识别受体(pattern recognition receptor,PRR)经由病原体相关分子模式(pathogen-associated molecular pattern,PAMP)和损伤相关分子模式(damage-associated molecular pattern,DAMP)进行识别并结合相应配体,组装形成胞浆内的多蛋白复合物,激活炎性Caspase-1和Caspase-4/5/11而进一步切割GSDMD蛋白对细胞膜进行打孔,促进细胞焦亡的发生。同时炎性小体作用于下游分子,促进炎症细胞因子(如IL-1β、IL-18、IL-6等)、趋化因子、黏附分子等的成熟并通过破裂的细胞膜释放到细胞外,募集并激活更多炎症细胞,放大局部和全身的炎症反应[4]。现有的研究表明,焦亡的激活途径分为由炎症小体激活Caspase-1的经典细胞焦亡途径和被胞质脂多糖(lipopolysaccharide,LPS)激活Caspase-4/5/11的非经典细胞焦亡途径。
3.1经典细胞焦亡途径
3.1.1炎性小体 炎性小体(inflammasome)是一类多蛋白复合物,主要类型包括NLRP3、NLRP1、NLRC4、AIM2等,其概念在2002年由Tschopp实验室[24]首次提出,可在宿主天然免疫反应中识别多种外源或内源性入侵的微生物、刺激性和损伤信号,与细胞死亡有密切的关系。炎性小体的基本结构主要有PRRs、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein containing CARD,ASC)和pro-caspase-1(CASP1)三部分[25,26]。其中,PRRs识别危险信号分子,细胞受到不同的信号刺激,相应的炎性小体会通过各种激活方式被激活发挥作用。ASC接头蛋白分子可通过其同源蛋白互作结构域PYD(pyrin domain,PYD)和C端半胱天冬酶募集域(C-terminal caspase recruitment domain,CARD)的寡聚化来募集上下游的PRRs和Pro-caspase-1,组装成炎性小体进行下游的信号转导。ASC不是NLRP1和NLRC4组装炎性小体的必要结构,但在一定程度上可以协助NLRP1和NLRC4易于Caspase-1的活化[27]。Caspase-1作为效应分子,在细胞中以无活性的酶原状态Pro-caspase-1存在,被ASC招募激活,活化的Caspase-1剪切促炎因子前体Pro-IL-1β和Pro-IL-18,促进IL-1β和IL-18的成熟与释放,招募更多的炎症细胞聚集,扩大炎症反应,提高机体先天性免疫防御能力,达到保护宿主细胞的目的[28]。
NLRP3是目前为止研究最深入的炎性体,由PYD、核苷酸结合寡聚化结构域(nucleotide binding oligomerization domain,NOD),亮氨酸富集重复序列(leucine-rich repeats,LRR)和CARD结构域构成,可被大多数病毒、细菌、真菌的DNA或RNA、细菌穿孔素、紫外线等外源性危险信号分子和ATP、晶体等内源性危险信号分子所激活[29-31],在该过程中,NEK7作为一种与有丝分裂纺锤体形成和中心体分离密切相关的重要蛋白质,能够调节平衡细胞分裂和炎性体活性,可激活NLRP3以应答经典和非经典刺激。NLRP1是第一个被报道可形成炎性复合物的NOD样受体(NLRs),人类仅具有一种NLRP1蛋白,但小鼠NLRP1蛋白具有多态性,编码3个旁系同源物:NLRP1a,1b和1c。人NLRP1含有N末端PYD,NOD,LRR,FIIND结构域(domain with function to find,FIIND)和C末端的CARD结构域,而小鼠NLRP1分子缺乏PYD[29]。在FIIND结构域的自蛋白水解作用下,含有保护性抗原和致死因子的炭疽芽孢杆菌毒素可通过N-末端接头区域诱导激活NLRP1炎性体,通过ASC依赖性募集激活Caspase-1形成炎性体复合物或通过CARD-CARD相互作用与Caspase-1直接结合。 NLRC4的初步研究表明其与凋亡蛋白酶激活因子(APAF1)有一定的相似性,包含LRR、NOD、CARD结构域[32],主要受革兰氏阴性菌的刺激而激活,致病菌如伤寒沙门氏菌亚种T3SS将效应蛋白释放到细胞溶胶中,这些致病相关蛋白被NAIPs家族识别,再进一步招募NLRC4来组装形成炎症小体复合物。AIM2是双链DNA(dsDNA)的细胞溶质受体,由PYD和具有200个氨基酸重复的结构域(HIN200)组成[29,33],HIN结构域的带正电荷的表面与DNA结合,PYD募集ASC以组装炎性体复合物。
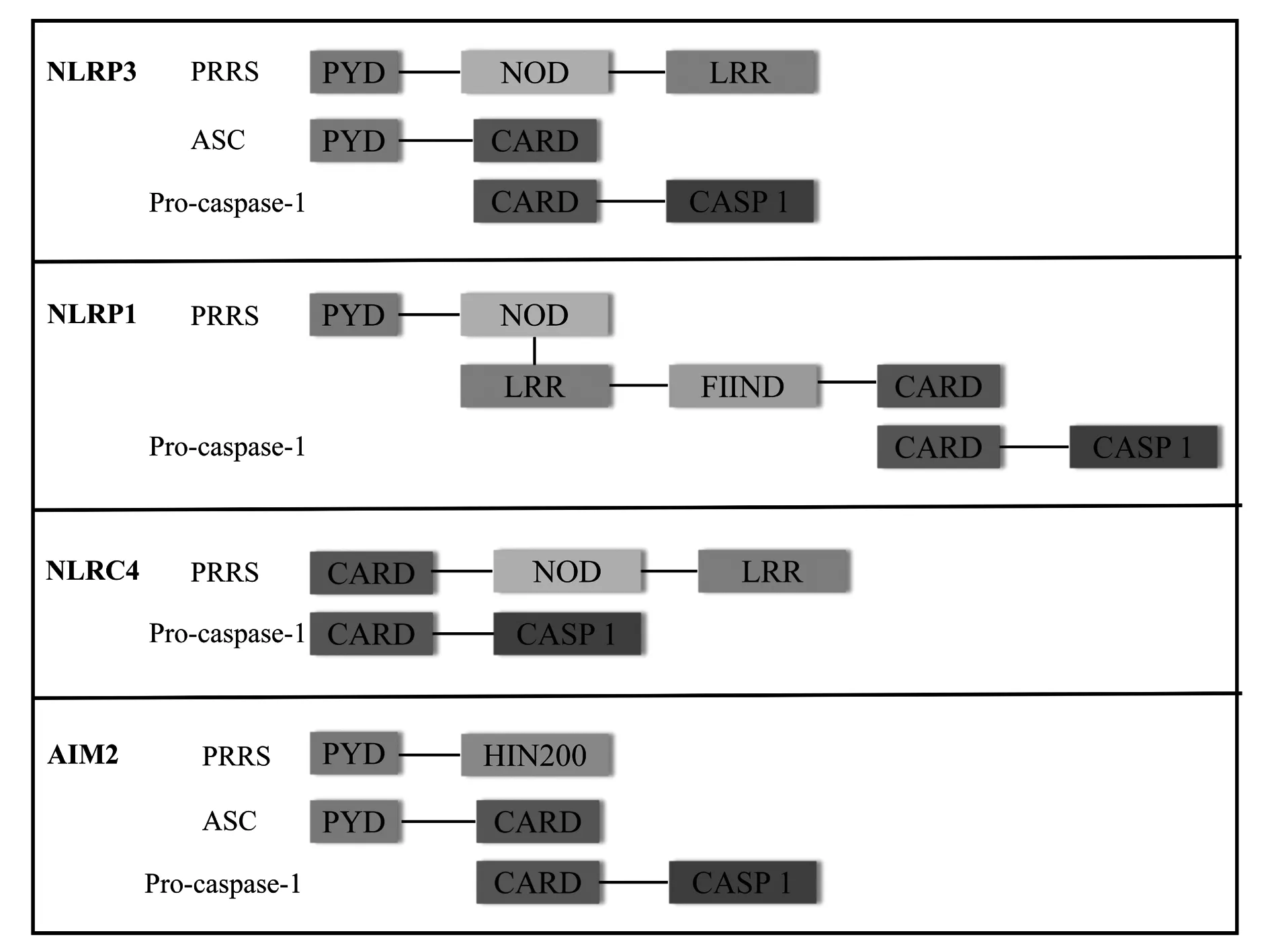
图1 炎性小体结构示意图Fig.1 Structure schematic diagram of inflammasomes
3.1.2炎性小体激活经典细胞焦亡通路 经典细胞焦亡途径的激活主要依赖于PRRs接受危险信号分子刺激,通过ASC招募Pro-caspase-1组装形成炎性小体,激活Caspase-1分子进一步切割下游GSDMD目的蛋白,促进细胞焦亡的发生。新的研究表明,GSDMD蛋白是炎性 Caspase的共同底物,是细胞焦亡的效应执行蛋白,形象地被称为“杀手蛋白”,在两条焦亡通路中均扮演着关键的角色。在细胞内环境中,GSDMD蛋白存在于细胞质中,可接受经典焦亡途径中被激活的Caspase-1和非经典焦亡途径中被激活的Caspase-4/5/11刺激,在特定位点将GSDMD蛋白分割成亲脂性N端结构域和亲水性C端结构域,其中N端结构域可与生物膜结合,在膜内侧聚集形成孔道,水分子侵入细胞,触发细胞焦亡[34],如图2所示。炎性小体的激活和GSDMD蛋白在细胞焦亡中起着十分重要的作用,其具体的激活信号通路之间并不是独立发挥作用,而是相互联系和影响,其过程还有待深入研究。
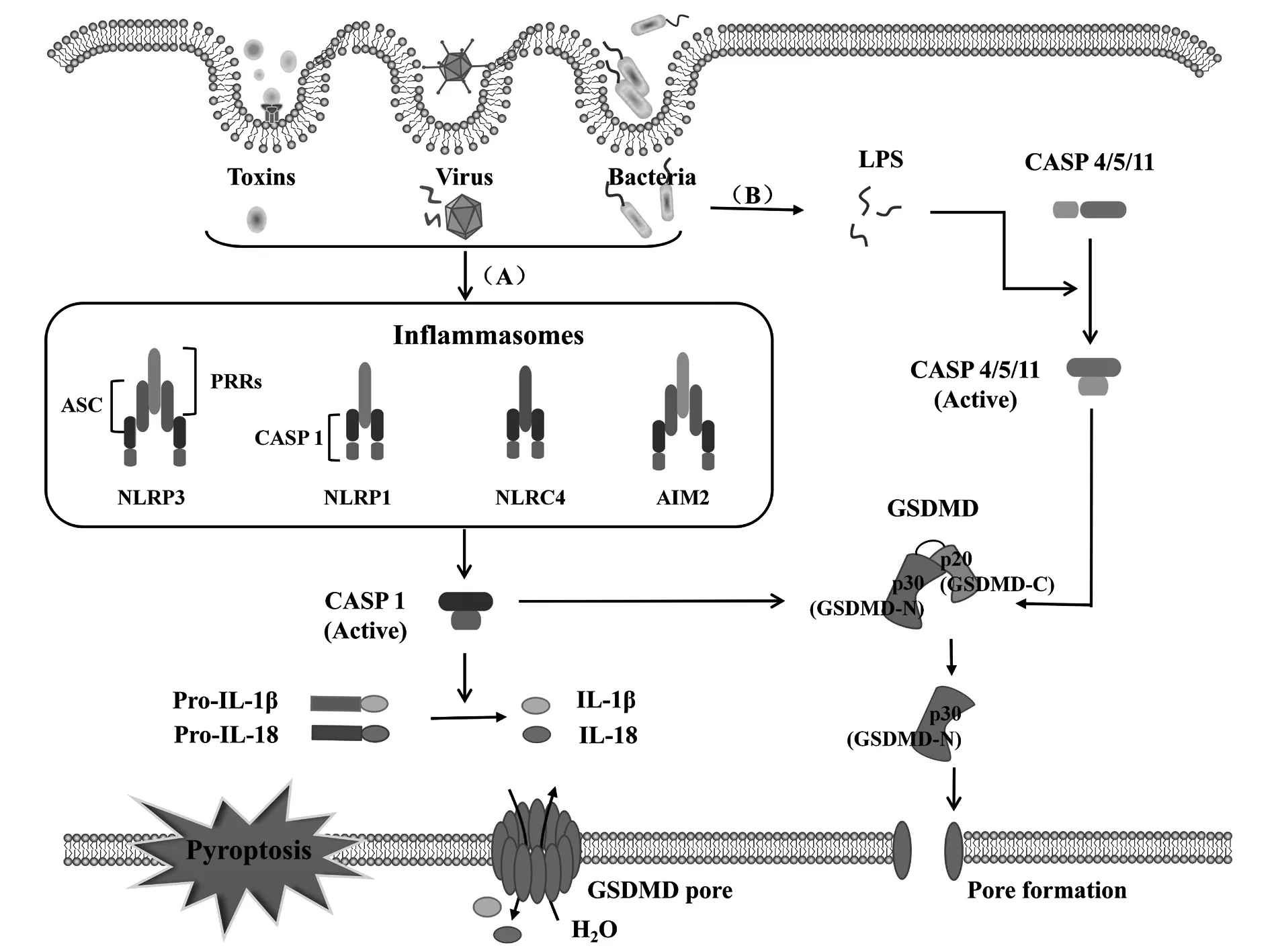
图2 经典焦亡途径与非经典焦亡途径Fig.2 Canonical pyroptosis pathway and noncanonical pyroptosis pathway
3.2非经典细胞焦亡途径 大量研究发现,细胞中还存在非依赖Caspase-1的细胞焦亡途径,与经典途径不同,非经典焦亡途径依赖于Caspase-4/5/11的激活。在受到胞质内LPS刺激后,小鼠Caspase-11和它的人类对应物Caspase-4/5可与LPS的保守结构脂质A直接结合被激活,活化的Caspase-4/5/11进一步切割GSDMD蛋白并促进细胞焦亡的发生[35,36],如图2所示。随着研究的不断深入细化,一些新的分子机制逐渐被发现。Pannexin-1是细胞膜上的通道蛋白,控制小分子物质进出,在非经典焦亡LPS进行免疫应答的过程中发挥重要作用,Caspase-11的激活会导致Pannexin-1蛋白断裂形成通路,将ATP释放到细胞外,当P2X7受体受到ATP长期反复的刺激时,P2X7通道开放使K+、Na+、Ca2+外流,打破细胞膜内外离子平衡,细胞发生渗透性肿胀进而膜破裂,最终导致细胞死亡[37]。
4 相关疾病与展望
大量研究和数据表明细胞焦亡普遍涉及感染性疾病、神经性疾病、动脉粥样硬化、自身免疫性等多种疾病的发生转归过程中,并且在癌症的发展进程中也发挥重要作用。早期细胞焦亡的发现和初步研究都是建立在细菌感染的模型基础上,在弗氏志贺氏杆菌、李斯特杆菌等细菌被证实可诱导细胞发生焦亡后[38-41],近年来也接连验证了肺炎链球菌[42]、耐药性金黄色葡萄球菌[43]及免疫缺陷类病毒[44]等感染机体可激活Caspase-1,并诱导发生细胞焦亡,引发多种感染性疾病。近年来神经系统疾病的研究中也发现脑缺血、脑损伤、帕金森症、阿尔兹海默症等疾病均与Caspase-1介导的经典细胞焦亡有关,涉及IL-1β和IL-18的表达升高和炎症反应的发生[45]。研究已经证实,通过沉默NLRP1或阻断Caspase-1的信号转导可以对疾病的发生发展起明显的抑制作用,因此认为Caspase-1是感染性和神经系统疾病的重要靶点。细胞焦亡在动脉粥样硬化(atherosclerosis,AS)和动脉粥样硬化斑块的形成中起促进作用,体外实验验证了[46]血浆中氧化型低密度脂蛋白(oxidized low density lipoprotein,oxLDL)可以引起细胞中脂质堆积沉降,促使巨噬细胞胆固醇化,激活NLRP3释放Caspase-1分子诱导细胞焦亡发生,引发并放大炎症反应的同时加速了动脉粥样硬化,若对NLRP3进行基因沉默阻断其信号途径,可使病情得到控制,起保护作用。当宿主免疫系统应答出现异常时,细胞焦亡引起的炎症级联反应吸引更多的免疫细胞到这一区域,引起恶性循环造成对免疫系统严重的破坏,引发类风湿性关节炎、系统性红斑狼疮(SLE)等自身免疫性疾病,相关研究表明通过抑制相应病原体激活的炎症小体,可使Caspase-1表达下调,降低IL-1β和IL-18的血清表达水平,有效改善疾病的恶化情况[47,48]。一直以来,以艾滋病为首的免疫系统缺陷类疾病都没有有效的治疗方案,如何抑制受感染的T细胞发生焦亡并提高机体免疫力会对这类疾病的预防和治疗提供新的思路和更有效的方法。Gasdermins家族蛋白广泛参与到肝癌、胃癌、乳腺癌等多种肿瘤的发生发展中,研究人员指出GSDME诱导细胞焦亡具有可调性[19-21],针对肿瘤细胞的免疫原性可采用特异性的药物进行治疗,为化疗耐药提供了新思路,更具体的作用机制和怎样提高治疗效果并减少对患者的毒副作用亟待更深入细致的研究。
焦亡是一种新的程序性炎症死亡,其经典通路由Caspase-1依赖性介导,通过炎性小体和Caspase-1的结合、激活、释放细胞内容物,非经典炎性体通路由Caspase-4/5/11介导,通过LPS激活Caspase-4/5/11诱发细胞焦亡[49]。炎性体生物学处于不断发展中,细胞死亡的机制扩展了关于炎性体的现有知识。然而,还有许多模式识别受体的功能和特异性配体仍然未知,PRRs激活和细胞稳态之间的相互作用及其对生理功能的影响仍在研究中,了解受体激活机制和配体识别的受体结构对于设计新的免疫疗法将是至关重要的。细胞焦亡作为机体天然免疫反应理论研究、多种炎症和免疫性相关疾病的模型逐渐成为科研工作者们关注的焦点,与机制研究较为完整的细胞凋亡相比,还存在很多尚未阐明的关键科学问题。细胞焦亡作为一种病理性的细胞主动裂解性死亡方式,一方面,焦亡的发生有助于消除病原体并防止感染,另一方面,过量的Caspase激活可导致级联炎症反应。细胞焦亡与多种疾病的发生发展密切相关,进一步研究Gasdermins家族的调控机制与相关疾病发生转归的关系,将对疾病的预防诊断、临床新型药物的研发提供更直接有效的帮助。
